21/02/2022
Fremstillingen af lægemidler er en af de mest regulerede processer i verden, og med god grund. Når det kommer til lægemidler, der stadig er under udvikling – de såkaldte forsøgslægemidler – bliver kravene endnu mere specifikke og komplekse. Disse produkter, som er kernen i kliniske forsøg, er afgørende for medicinsk innovation, men deres produktion indebærer unikke udfordringer. De er ofte ikke fuldt karakteriserede, produceres i små partier og kan undergå ændringer i løbet af udviklingsfasen. Derfor er overholdelse af principperne for God Fremstillingspraksis (Good Manufacturing Practice, GMP) ikke bare en formalitet, men en absolut nødvendighed for at sikre patienternes sikkerhed og integriteten af de indsamlede data.

Hvad er et Forsøgslægemiddel?
Et forsøgslægemiddel er en farmaceutisk form af et aktivt stof eller placebo, der testes eller anvendes som reference i et klinisk forsøg. Dette inkluderer også produkter, der allerede har en markedsføringstilladelse, men som anvendes eller sammensættes på en anden måde end den godkendte, eller når det bruges til en ikke-godkendt indikation. Formålet med at anvende disse lægemidler i kliniske forsøg er at indsamle afgørende data om deres sikkerhed og effekt, før de potentielt kan blive godkendt til almen brug. I de tidlige udviklingsfaser er viden om produktets egenskaber, såsom stabilitet og optimale produktionsmetoder, ofte begrænset. Dette adskiller dem markant fra kommercielt tilgængelige lægemidler, der fremstilles i store, veletablerede og validerede processer.
Principperne i God Fremstillingspraksis (GMP)
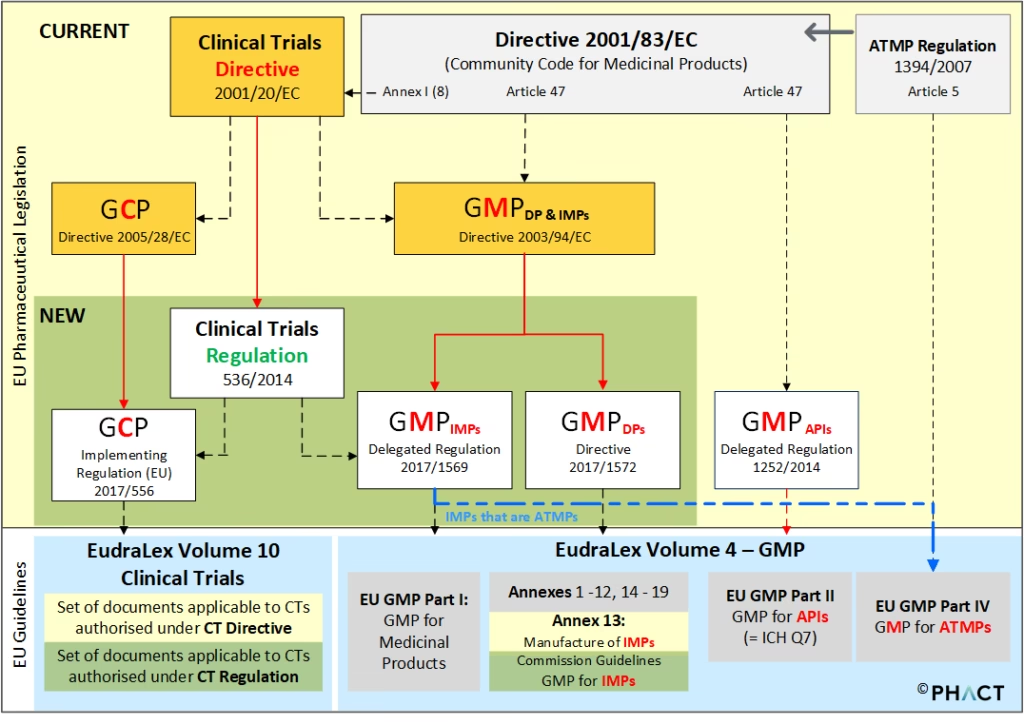
God Fremstillingspraksis er et kvalitetssikringssystem, der sikrer, at lægemidler konsekvent produceres og kontrolleres i henhold til de kvalitetsstandarder, der er passende for deres tilsigtede anvendelse. I Den Europæiske Union er disse principper og retningslinjer specificeret i 'The Rules Governing Medicinal Products in The European Community, Volume IV'. For forsøgslægemidler gælder et specifikt anneks (Anneks 13), der adresserer de særlige forhold, der gør sig gældende for disse produkter.
Hovedformålet med GMP er at minimere de risici, der er forbundet med farmaceutisk produktion, som ikke kan elimineres gennem testning af det endelige produkt. De primære risici er:
- Krydskontaminering (forurening med uønskede stoffer).
- Forvekslinger (f.eks. forkert mærkning).
- Produktion af et produkt, der ikke lever op til de fastsatte specifikationer.
Ved at implementere et robust GMP-system sikrer man, at hver batch af et forsøgslægemiddel har den samme kvalitet, styrke og renhed, som er defineret i forsøgsprotokollen. Dette er altafgørende for, at de data, der genereres i det kliniske forsøg, er pålidelige og reproducerbare.
Særlige Udfordringer og GMP-tilpasninger
Produktionen af forsøgslægemidler adskiller sig fra rutinemæssig kommerciel produktion på flere punkter, hvilket kræver en fleksibel, men kontrolleret tilgang til GMP.
Produktionsprocessen
Processen er sjældent veletableret. Der kan forekomme hyppige ændringer i formulering, doseringsstyrke eller fremstillingsmetode, efterhånden som mere viden opnås. GMP kræver, at alle ændringer er veldokumenterede og evalueret for deres potentielle indvirkning på produktets kvalitet og sikkerhed. Procesvalidering, som for kommercielle produkter er en forudsætning for frigivelse, udføres ofte sideløbende med den kliniske udvikling for forsøgslægemidler.
Dokumentation
En af hjørnestenene i GMP er Dokumentation. For forsøgslægemidler er dette endnu mere kritisk. Der skal være en komplet sporbarhed fra råmaterialer til det færdige produkt, der administreres til en patient. Specifikationer for råvarer og det færdige produkt udvikles løbende. Produktspecifikationsfilen (Product Specification File) er et centralt dokument, der indeholder eller henviser til al information, der er nødvendig for at frigive en batch.

Mærkning og Blinding
Mærkningen af forsøgslægemidler er ekstremt kompleks. Etiketterne skal indeholde en lang række oplysninger for at sikre korrekt anvendelse og sporbarhed (f.eks. protokolnummer, patient-ID, udløbsdato), men uden at afsløre produktets identitet, hvis forsøget er blindet. Blinding er processen, hvor man skjuler, om patienten modtager det aktive lægemiddel eller placebo. Dette er afgørende for at undgå bias i forsøgsresultaterne. GMP-systemet skal sikre, at emballerings- og mærkningsprocesserne er fejlfrie for at opretholde blindingen og garantere Patientsikkerhed.
Den Kvalificerede Persons Rolle
I EU skal hver batch af et lægemiddel, inklusive forsøgslægemidler, certificeres og frigives af en Kvalificeret Person (QP - Qualified Person). QP'en har det personlige juridiske ansvar for at sikre, at batchen er fremstillet og kontrolleret i overensstemmelse med GMP og de relevante regulatoriske krav og specifikationer. For forsøgslægemidler er denne rolle særligt krævende, da QP'en skal vurdere batcher baseret på et informationsgrundlag, der er under konstant udvikling.
Sammenligning: Forsøgslægemiddel vs. Kommercielt Lægemiddel
For at illustrere forskellene kan en sammenligning være nyttig:
| Karakteristikum | Kommercielt Lægemiddel | Forsøgslægemiddel |
|---|---|---|
| Batchstørrelse | Stor og ensartet | Lille og ofte varierende |
| Produktionsproces | Fuldt valideret og fastlåst | Under udvikling, kan ændres |
| Produktviden | Omfattende (stabilitet, bivirkn.) | Begrænset og under opbygning |
| Mærkning | Standardiseret | Kompleks, forsøgsspecifik, ofte blindet |
| Formål | Behandling af patienter | Indsamling af data om sikkerhed og effekt |
Ofte Stillede Spørgsmål (OSS)
Hvorfor er reglerne anderledes for forsøgslægemidler?
Reglerne er tilpasset for at håndtere den usikkerhed og de konstante ændringer, der er en naturlig del af lægemiddeludvikling. De giver den nødvendige fleksibilitet til at justere processer baseret på ny viden, samtidig med at der opretholdes strenge kontroller for at beskytte forsøgsdeltagerne.
Hvem kontrollerer, at GMP overholdes?
De nationale lægemiddelmyndigheder (i Danmark, Lægemiddelstyrelsen) er ansvarlige for at inspicere produktionsfaciliteter for at sikre, at de overholder GMP-reglerne. En virksomhed skal have en fremstillingstilladelse for at kunne producere forsøgslægemidler.
Hvad sker der, hvis et forsøgslægemiddel ikke er produceret efter GMP?
Hvis et produkt ikke overholder GMP, kan det udgøre en alvorlig risiko for patienterne. Data indsamlet ved brug af produktet vil sandsynligvis blive anset for upålidelige af myndighederne, hvilket kan føre til, at hele det kliniske forsøg bliver afvist. I sidste ende kan det forhindre et potentielt vigtigt lægemiddel i at nå markedet.
Konklusion: En Balanceakt for Sikkerhed og Innovation
Fremstillingen af forsøgslægemidler er en kompleks disciplin, der balancerer behovet for videnskabelig fleksibilitet med det ufravigelige krav om patientsikkerhed og dataintegritet. Principperne i God Fremstillingspraksis udgør det fundament, der gør denne balance mulig. Ved at sikre, at hvert enkelt hætteglas, tablet eller injektion, der anvendes i kliniske forsøg, er produceret under kontrollerede og veldokumenterede forhold, bygger man bro mellem de tidlige stadier i laboratoriet og den endelige godkendelse af et nyt, sikkert og effektivt lægemiddel til gavn for patienter verden over.
Hvis du vil læse andre artikler, der ligner GMP-regler for Forsøgslægemidler, kan du besøge kategorien Farmaci.