19/12/2025
Når du tager medicin, hvad enten det er en simpel hovedpinetablet eller en avanceret kræftbehandling, gør du det med en grundlæggende tillid til, at produktet er sikkert og virker som det skal. Men hvem garanterer denne sikkerhed på tværs af et helt kontinent? Svaret ligger hos Det Europæiske Lægemiddelagentur, bedre kendt som EMA. Denne organisation er hjørnestenen i Europas system til regulering af lægemidler og spiller en afgørende rolle i at beskytte og fremme folke- og dyresundheden i hele Den Europæiske Union (EU). I denne artikel dykker vi ned i, hvad EMA er, hvordan det fungerer, og hvorfor dets arbejde er så vitalt for os alle.

Hvad er Det Europæiske Lægemiddelagentur (EMA)?
Det Europæiske Lægemiddelagentur blev oprettet i 1995, oprindeligt under navnet Det Europæiske Agentur for Lægemiddelvurdering (EMEA), og har siden udviklet sig til at være den centrale videnskabelige autoritet for lægemidler i Europa. Dets primære mission er at beskytte og fremme folke- og dyresundheden gennem grundig evaluering og overvågning af lægemidler. EMA dækker de 27 EU-medlemslande samt Island, Liechtenstein og Norge, som tilsammen udgør Det Europæiske Økonomiske Samarbejdsområde (EØS).
I modsætning til hvad mange tror, er EMA ikke en enkeltstående, monolitisk enhed, der træffer alle beslutninger alene. Det fungerer snarere som et netværk, der koordinerer de videnskabelige ressourcer fra de nationale lægemiddelmyndigheder i medlemslandene. Dette skaber et robust system, hvor den bedste ekspertise fra hele Europa samles for at foretage en videnskabelig vurdering af lægemidler. EMA's ansvar omfatter både lægemidler til mennesker og dyr, men ikke fødevarer, som håndteres af Den Europæiske Fødevaresikkerhedsautoritet (EFSA).
En af EMA's vigtigste funktioner er den "centraliserede procedure", som giver en virksomhed mulighed for at indsende en enkelt ansøgning om markedsføringstilladelse for at få den vurderet af EMA. Hvis agenturet anbefaler en godkendelse, kan Europa-Kommissionen udstede én enkelt tilladelse, der er gyldig i alle EU/EØS-lande. Dette er især vigtigt for innovative lægemidler, f.eks. inden for bioteknologi, kræftbehandling og sjældne sygdomme.
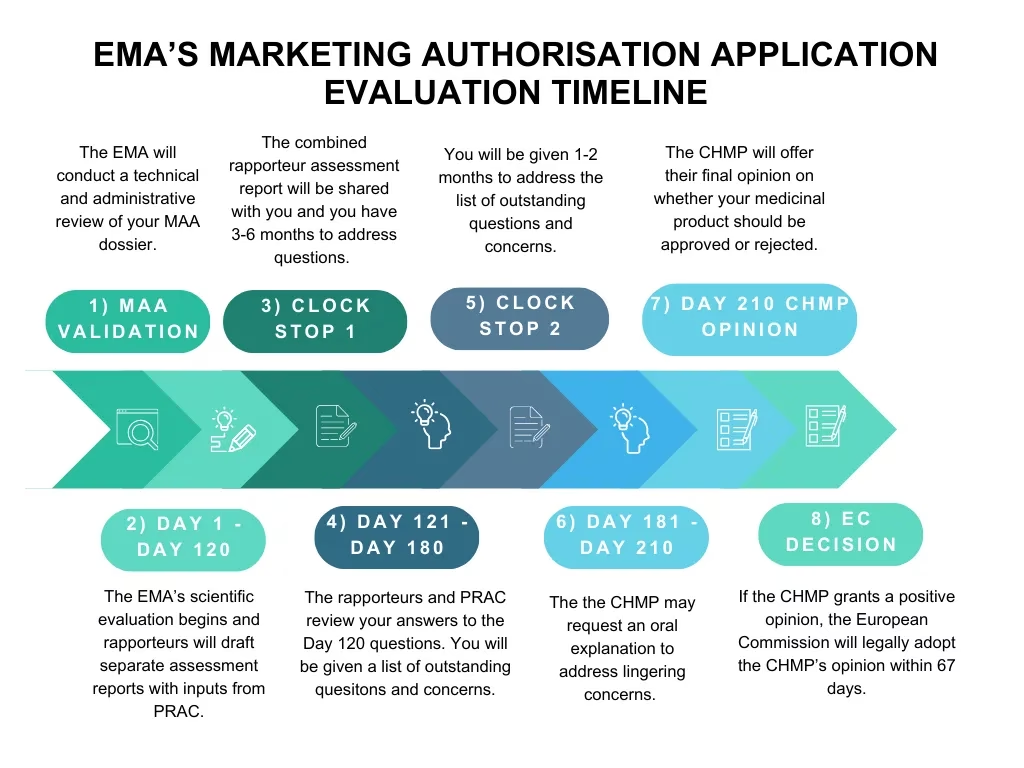
Hvordan fungerer godkendelsesprocessen? CHMP's centrale rolle
Kernen i EMA's videnskabelige arbejde for humane lægemidler er Udvalget for Lægemidler til Mennesker (CHMP). Dette udvalg består af eksperter fra alle medlemslande og er ansvarligt for at vurdere data om et nyt lægemiddels kvalitet, sikkerhed og effekt. Processen er bygget op omkring principperne om peer review og kollegial beslutningstagning for at sikre den højest mulige videnskabelige standard.
Processen for en ny ansøgning foregår typisk således:
- Udnævnelse af rapportører: For hver ny ansøgning udpeges to medlemmer af CHMP fra forskellige lande som henholdsvis "rapportør" og "co-rapportør". Deres opgave er at lede den videnskabelige vurdering uafhængigt af hinanden. Dette sikrer en objektiv og mangesidet evaluering.
- Vurderingshold: Hver rapportør samler et hold af eksperter fra deres nationale myndighed. Disse hold gennemgår ansøgerens data i dybden, analyserer resultater fra kliniske forsøg og identificerer eventuelle usikkerheder eller spørgsmål, som lægemiddelproducenten skal besvare.
- Peer Review: Udover de to hovedvurderinger udpeger CHMP også en eller flere "peer reviewers" blandt de øvrige udvalgsmedlemmer. Deres rolle er at granske vurderingsrapporterne kritisk og sikre, at den videnskabelige argumentation er solid, klar og robust.
- Plenarmøder: Alle CHMP-medlemmer gennemgår rapporterne og kommentarerne og diskuterer dem på månedlige plenarmøder. Gennem disse diskussioner, og efterhånden som ansøgeren leverer yderligere data eller afklaringer, formes udvalgets samlede holdning. Denne iterative proces sikrer, at den endelige anbefaling er baseret på en grundig og fælles analyse af alle tilgængelige beviser.
Resultatet af denne omfattende proces er en videnskabelig udtalelse om, hvorvidt fordelene ved lægemidlet opvejer risiciene. Denne udtalelse sendes derefter til Europa-Kommissionen, som træffer den endelige juridiske afgørelse.
Lægemiddelovervågning: Sikkerhed efter godkendelse
EMA's arbejde stopper ikke, når et lægemiddel er blevet godkendt. At sikre lægemidlers sikkerhed er en kontinuerlig proces, der varer hele produktets levetid. Dette kaldes lægemiddelovervågning eller "pharmacovigilance". Målet er konstant at overvåge og vurdere lægemidlers sikkerhedsprofil og gribe ind, hvis nye risici opdages.

Centrale elementer i EMA's lægemiddelovervågning omfatter:
- EudraVigilance-databasen: Dette er et avanceret europæisk system til indsamling og håndtering af bivirkningsrapporter. Både sundhedspersonale, patienter og lægemiddelvirksomheder kan indberette formodede bivirkninger. Databasen gør det muligt for EMA og de nationale myndigheder at analysere store mængder data for at opdage potentielle sikkerhedssignaler – dvs. nye eller ændrede risici forbundet med et lægemiddel.
- Risikostyringsplaner (RMP): For hvert nyt lægemiddel kræves en detaljeret risikostyringsplan. Denne plan beskriver lægemidlets kendte og potentielle risici og skitserer, hvordan disse risici vil blive overvåget og minimeret, når det kommer på markedet. Det er en proaktiv tilgang til sikkerhed.
- Periodiske sikkerhedsopdateringer (PSURs): Lægemiddelvirksomheder er forpligtet til regelmæssigt at indsende rapporter, der opsummerer alle nye sikkerhedsdata for deres produkter. Disse rapporter gennemgås af EMA for at vurdere, om benefit-risk-balancen for lægemidlet fortsat er positiv.
- Udvalget for Risikovurdering inden for Lægemiddelovervågning (PRAC): Dette specialiserede udvalg er ansvarligt for at vurdere alle aspekter af risikostyring for lægemidler til mennesker. PRAC analyserer sikkerhedssignaler og giver anbefalinger til CHMP om nødvendige tiltag, som kan omfatte alt fra en opdatering af indlægssedlen til en suspension eller tilbagetrækning af lægemidlet.
EMA's samspil med andre EU-institutioner
EMA er en videnskabelig instans, men det opererer inden for en bredere politisk og juridisk ramme fastsat af EU's centrale institutioner. Samarbejdet mellem disse organer sikrer en klar adskillelse mellem videnskabelig vurdering og politisk beslutningstagning.
Rollerne i lægemiddelregulering
| Institution | Primær Rolle i Lægemiddelregulering |
|---|---|
| EMA (CHMP/PRAC) | Udfører den uafhængige videnskabelige vurdering af et lægemiddels kvalitet, sikkerhed og effekt og afgiver en anbefaling. |
| Europa-Kommissionen | Træffer den endelige, juridisk bindende afgørelse om markedsføringstilladelse baseret på EMA's videnskabelige anbefaling. |
| Europa-Parlamentet | Vedtager lovgivning om lægemidler sammen med Rådet og fører tilsyn med EMA's arbejde (især gennem ENVI-udvalget). |
| Rådet for Den Europæiske Union | Vedtager lovgivning om lægemidler sammen med Parlamentet. Består af medlemslandenes ministre. |
Dette system sikrer, at beslutninger om lægemidler er baseret på solid videnskab, samtidig med at de er forankret i en demokratisk og juridisk proces, der repræsenterer alle EU-borgere.
Ofte Stillede Spørgsmål
Hvad er forskellen på EMA og de nationale lægemiddelmyndigheder som f.eks. Lægemiddelstyrelsen i Danmark?
EMA og de nationale myndigheder udgør tilsammen det europæiske lægemiddelnetværk. Forskellen ligger primært i deres ansvarsområder. EMA er ansvarlig for den centraliserede godkendelsesprocedure, som er obligatorisk for visse typer af innovative lægemidler. De nationale myndigheder, som Lægemiddelstyrelsen, håndterer godkendelser af mange andre lægemidler, der kun skal markedsføres nationalt (via nationale procedurer eller gensidige anerkendelsesprocedurer). Desuden spiller de nationale myndigheder en afgørende rolle i at overvåge lægemidler på deres eget marked, inspicere producenter og indsamle bivirkningsrapporter, som de deler med EMA.

Kan en lægemiddelproducent selv vælge, om de vil søge godkendelse via EMA eller nationalt?
Det afhænger af lægemidlet. For de mest innovative lægemidler – såsom lægemidler fremstillet ved hjælp af bioteknologi, lægemidler mod kræft, diabetes, neurodegenerative sygdomme, HIV/AIDS og sjældne sygdomme – er den centraliserede procedure via EMA obligatorisk. Dette sikrer, at patienter i hele Europa får adgang til vigtige nye behandlinger samtidigt, og at disse lægemidler vurderes af den bredest mulige ekspertise. For andre lægemidler, f.eks. generika, kan producenten ofte vælge mellem den centraliserede procedure eller nationale procedurer.
Hvad sker der, hvis der opdages en alvorlig bivirkning ved et godkendt lægemiddel?
Hvis der opstår mistanke om en ny, alvorlig bivirkning, igangsættes en hurtig undersøgelsesproces. Rapporter om bivirkningen analyseres i EudraVigilance-databasen for at identificere et mønster (et "signal"). EMA's sikkerhedsudvalg, PRAC, vurderer signalet og alle tilgængelige data. Hvis risikoen bekræftes, kan PRAC anbefale en række tiltag. Det kan være en opdatering af produktinformationen og indlægssedlen for at advare læger og patienter, en begrænsning i hvem der må bruge lægemidlet, eller i de mest alvorlige tilfælde en suspension eller fuldstændig tilbagetrækning af lægemidlets markedsføringstilladelse i hele EU.
Det Europæiske Lægemiddelagentur er således meget mere end blot et administrativt organ. Det er et dynamisk og videnskabsdrevet netværk, der fungerer som Europas vogter af lægemiddelsikkerhed. Fra den første vurdering af et nyt, lovende lægemiddel til den livslange overvågning af dets sikkerhed på markedet, arbejder tusindvis af eksperter på tværs af kontinentet sammen under EMA's koordinering for at sikre, at den medicin, vi stoler på, er sikker, effektiv og af høj kvalitet.
Hvis du vil læse andre artikler, der ligner EMA: Sådan sikres din medicin i Europa, kan du besøge kategorien Sundhed.