
18/07/2012
Rejsen for et nyt lægemiddel, fra den spæde idé i et forskningslaboratorium til det færdige produkt på apotekets hylde, er en af de mest komplekse, tidskrævende og dyre processer i den moderne verden. Det er en fortælling om videnskabelig nysgerrighed, omhyggelige tests, strenge regulativer og et konstant håb om at kunne lindre symptomer, helbrede sygdomme og forbedre livskvaliteten for millioner af mennesker. Mens vi ofte tager en pille uden at tænke nærmere over det, ligger der i gennemsnit 12-15 års intensivt arbejde og en investering på flere milliarder kroner bag hvert enkelt godkendt præparat. Denne artikel vil guide dig gennem de afgørende faser i denne utrolige proces og belyse de udfordringer og innovationer, der former fremtidens medicin.
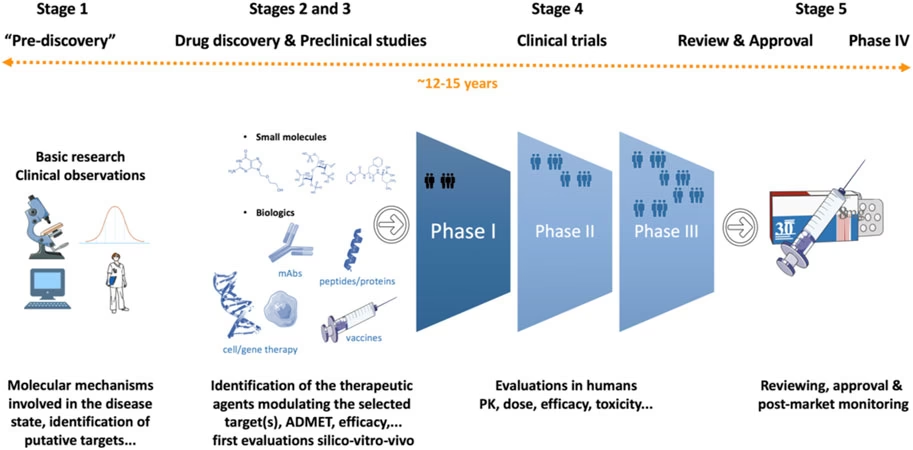
De fem hovedfaser i udviklingen af et lægemiddel
Processen med at opdage og udvikle et nyt lægemiddel kan overordnet inddeles i fem centrale etaper. Hver etape har sit eget formål og involverer et væld af eksperter fra forskellige discipliner, fra biologer og kemikere til læger og statistikere.
1. For-opdagelsesfasen: Grundforskning og identifikation af mål
Alt begynder med en dybdegående forståelse af en sygdom. I denne indledende fase, som ofte foregår på universiteter og i akademiske forskningsmiljøer, forsøger forskere at afdække de biologiske mekanismer, der ligger bag en lidelse. Hvilke proteiner, gener eller signalveje i kroppen er ude af balance? Målet er at identificere et såkaldt 'terapeutisk mål' (ofte et specifikt protein), som et potentielt lægemiddel kan påvirke for at genoprette kroppens normale funktion eller i det mindste lindre symptomerne. At finde det rigtige mål er ekstremt udfordrende og afgørende for hele projektets succes.
2. Opdagelsesfasen: Jagten på det aktive molekyle
Når et mål er identificeret, starter jagten på et molekyle, der kan interagere med det på den ønskede måde. Her screenes tusindvis, ja endda millioner, af kemiske forbindelser for at finde dem, der viser den rette aktivitet. Dette kan ske gennem high-throughput screening i laboratoriet eller ved hjælp af computer-assisterede metoder, hvor man virtuelt tester enorme biblioteker af molekyler. De mest lovende kandidater, kaldet 'hits' og senere 'leads', udvælges til videre optimering for at forbedre deres effektivitet og reducere potentielle bivirkninger.
3. Præklinisk udvikling: Sikkerhedstest før forsøg på mennesker
Før et lægemiddelkandidat kan testes på mennesker, skal det gennemgå en omfattende præklinisk fase. Her undersøges molekylets virkningsmekanisme i detaljer, og dets sikkerhedsprofil vurderes grundigt. Der udføres tests in vitro (i reagensglas og på cellekulturer) og in vivo (i levende organismer, typisk dyremodeller) for at undersøge toksicitet (giftighed), og hvordan stoffet optages, fordeles, metaboliseres og udskilles af kroppen (ADME-studier). Kun de få kandidater, der viser sig at være både effektive og tilstrækkeligt sikre i disse tests, kan gå videre til kliniske forsøg.
4. Kliniske forsøg: Test på mennesker
Dette er den længste og mest omkostningstunge del af processen, hvor lægemiddelkandidaten for første gang testes i mennesker. De kliniske forsøg er opdelt i flere faser:
- Fase I: Her testes lægemidlet på en lille gruppe (20-80) raske, frivillige forsøgspersoner. Hovedformålet er at vurdere sikkerhed, tolerabilitet og finde en sikker dosis.
- Fase II: Lægemidlet gives nu til en større gruppe patienter (100-500), der lider af den pågældende sygdom. Her undersøges det, om lægemidlet har den ønskede terapeutiske effekt, og man fortsætter med at indsamle sikkerhedsdata for at finde den optimale dosis. Mange kandidater falder fra i denne fase på grund af manglende effekt eller uacceptable bivirkninger.
- Fase III: Dette er store, ofte globale, studier med tusindvis af patienter (1.000-5.000). Formålet er endegyldigt at bevise lægemidlets effekt i forhold til eksisterende behandlinger eller placebo (snydemedicin). Studierne er typisk randomiserede og dobbeltblindede, hvilket betyder, at hverken patient eller læge ved, hvem der modtager det aktive stof. Disse studier er afgørende for at få myndighedernes godkendelse.
5. Godkendelse og overvågning efter markedsføring
Efter vellykkede fase III-studier samles al dokumentation i en omfattende ansøgning, som sendes til sundhedsmyndighederne, f.eks. Det Europæiske Lægemiddelagentur (EMA). Myndighederne gennemgår alle data for at vurdere lægemidlets kvalitet, sikkerhed og effekt. Hvis fordelene vurderes at opveje risiciene, bliver lægemidlet godkendt til markedsføring. Men arbejdet stopper ikke her. I den såkaldte fase IV, eller farmakovervågning, overvåges lægemidlet fortsat for at opdage sjældne eller langsigtede bivirkninger, som måske ikke blev opdaget i de kliniske forsøg.
Små molekyler vs. biologiske lægemidler: To verdener af medicin
Moderne lægemidler kan groft inddeles i to hovedkategorier: små molekyler og biologiske lægemidler. De adskiller sig markant i størrelse, fremstilling og ofte også pris.
Små molekyler er kemisk syntetiserede stoffer med lav molekylvægt, som vi kender fra traditionelle piller som f.eks. paracetamol eller ibuprofen. De kan typisk tages oralt og absorberes let i kroppen. Biologiske lægemidler er derimod store, komplekse molekyler, såsom proteiner, antistoffer eller vacciner, der er produceret i levende celler. På grund af deres størrelse og følsomhed skal de ofte gives som en injektion eller infusion.
Sammenligningstabel
| Egenskab | Små molekyler | Biologiske lægemidler |
|---|---|---|
| Struktur | Simpel, veldefineret kemisk struktur | Stor, kompleks struktur (f.eks. proteiner) |
| Fremstilling | Kemisk syntese | Produceret i levende celler (bioteknologi) |
| Administration | Ofte oral (piller, kapsler) | Typisk injektion eller infusion |
| Stabilitet | Generelt stabil | Følsom over for varme og nedbrydning |
| Omkostninger | Lavere produktionsomkostninger | Meget høje produktionsomkostninger |
Innovationer der fremskynder processen
Den traditionelle udviklingsproces er langsom og dyr med en meget høj fejlrate. Derfor arbejdes der konstant på nye metoder, der kan gøre processen mere effektiv.
Genanvendelse af lægemidler (Drug Repurposing)
En smart strategi er at undersøge, om eksisterende, godkendte lægemidler kan bruges til at behandle andre sygdomme, end de oprindeligt var tiltænkt. Da sikkerhedsprofilen for disse lægemidler allerede er kendt, kan man potentielt springe de tidlige udviklingsfaser over. Et berømt eksempel er thalidomid, der oprindeligt var et sovemiddel, men som efter at være blevet trukket tilbage på grund af alvorlige fosterskader, senere blev godkendt til behandling af spedalskhed og knoglemarvskræft.
Kunstig Intelligens (AI): Håb og realiteter
Kunstig intelligens er ved at revolutionere mange dele af lægemiddeludviklingen. AI-algoritmer kan analysere enorme mængder biologiske data for at identificere nye terapeutiske mål, designe nye molekyler med specifikke egenskaber og forudsige, hvilke patienter der vil have gavn af en given behandling. Selvom AI har potentialet til at gøre processen hurtigere og mere præcis, er teknologien stadig under udvikling. Kvaliteten af de data, AI-modellerne trænes på, er afgørende, og alle forudsigelser skal stadig verificeres grundigt gennem eksperimentelle tests. Princippet er: stol på AI, men verificer resultaterne.
Fremtiden: Mod personlig og effektiv medicin
Fremtiden for lægemiddeludvikling peger i retning af mere personlig medicin**, hvor behandlingen skræddersys til den enkelte patients genetiske profil og sygdomsmekanismer. Samtidig er der et stigende fokus på at udvikle alternativer til dyreforsøg, blandt andet ved hjælp af avancerede cellemodeller og 'organ-on-a-chip'-teknologier. Nye regulatoriske rammer, som f.eks. FDA Modernization Act 2.0 i USA, åbner allerede op for, at data fra sådanne metoder kan erstatte nogle dyreforsøg. Åbent samarbejde mellem industri, universiteter og non-profit organisationer bliver også stadig vigtigere for at dele viden og fremskynde udviklingen af medicin til patienter, der har mest brug for det.
Ofte Stillede Spørgsmål (OSS)
Hvorfor tager det så lang tid at udvikle et nyt lægemiddel?
Det tager lang tid på grund af de strenge krav til sikkerhed og effekt. Hver fase, fra grundforskning til de omfattende kliniske forsøg og myndighedsgodkendelse, kræver omhyggelig dataindsamling og analyse for at sikre, at lægemidlet er sikkert og virker som forventet, før det kan gives til patienter.
Hvad er forskellen på et 'lille molekyle' og et 'biologisk lægemiddel'?
Et 'lille molekyle' er en simpel kemisk forbindelse, der typisk kan laves i et laboratorium og tages som en pille. Et 'biologisk lægemiddel' er et stort, komplekst molekyle (f.eks. et antistof) produceret i levende celler, som normalt skal injiceres.
Kan kunstig intelligens erstatte forskere i lægemiddeludvikling?
Nej, ikke på nuværende tidspunkt. AI er et utroligt kraftfuldt værktøj, der kan assistere forskere ved at analysere data og generere hypoteser meget hurtigere end et menneske. Men den kritiske tænkning, kreativitet og eksperimentelle validering, som forskere bidrager med, er stadig uundværlig.
Hvorfor er nye lægemidler så dyre?
De høje omkostninger afspejler den enorme investering i forskning og udvikling over 10-15 år. For hvert lægemiddel, der bliver godkendt, er der tusindvis af kandidater, der er faldet fra undervejs. Prisen skal dække omkostningerne for alle disse fejlslagne forsøg samt de dyre fase III-studier.
Er dyreforsøg stadig nødvendige?
Historisk set har dyreforsøg været et lovkrav for at vurdere sikkerhed, før et lægemiddel må testes på mennesker. Der er dog et stigende globalt pres og videnskabelig udvikling i retning af at reducere og erstatte dyreforsøg med nye teknologier som avancerede cellekulturer og computermodeller, som bedre kan efterligne menneskelig biologi.
Hvis du vil læse andre artikler, der ligner Fra idé til apotek: Lægemidlets rejse, kan du besøge kategorien Sundhed.