13/10/2010
I den utroligt komplekse verden inde i vores celler findes der en gruppe molekylære arbejdsheste kendt som proteiner. De er ansvarlige for næsten alt, hvad der sker i vores krop – fra at transportere ilt i blodet og fordøje vores mad til at bekæmpe infektioner og overføre signaler i hjernen. Men for at et protein kan udføre sin specifikke opgave, skal det først folde sig sammen til en helt bestemt tredimensionel form. Denne 3D-struktur er altafgørende for dets funktion. Hvis vi kun kender proteinets 'ingrediensliste' – rækkefølgen af aminosyrer – hvordan kan vi så forudsige dets endelige, funktionelle form? Her kommer en kraftfuld bioinformatisk teknik ind i billedet: proteintrådning.
Hvad er Proteintrådning?
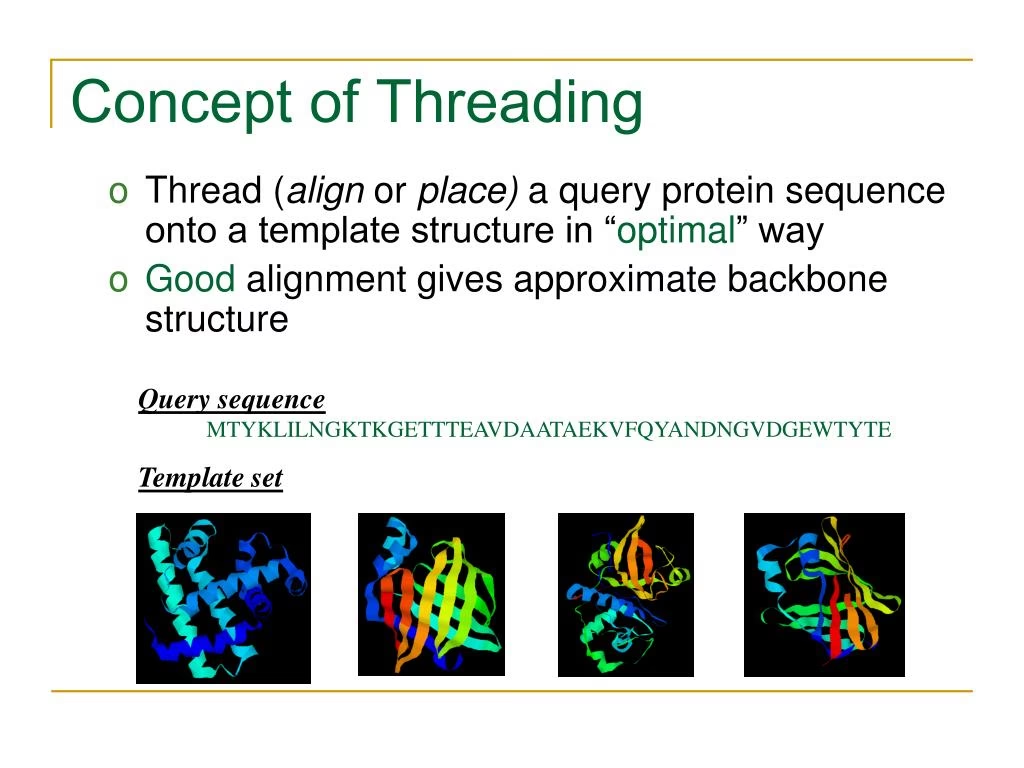
Forestil dig, at du har en lang snor med perler i forskellige farver og størrelser. Denne perlesnor repræsenterer en proteins aminosyresekvens. Forestil dig nu, at du har en samling af færdiglavede, indviklede skulpturer lavet af ståltråd. Disse skulpturer repræsenterer de kendte, grundlæggende proteinstrukturer, som forskere allerede har kortlagt. Proteintrådning, også kendt som 'fold recognition', er processen, hvor man prøver at 'tråde' sin perlesnor igennem hver af disse ståltrådsskulpturer for at se, hvilken skulptur den passer bedst ind i.
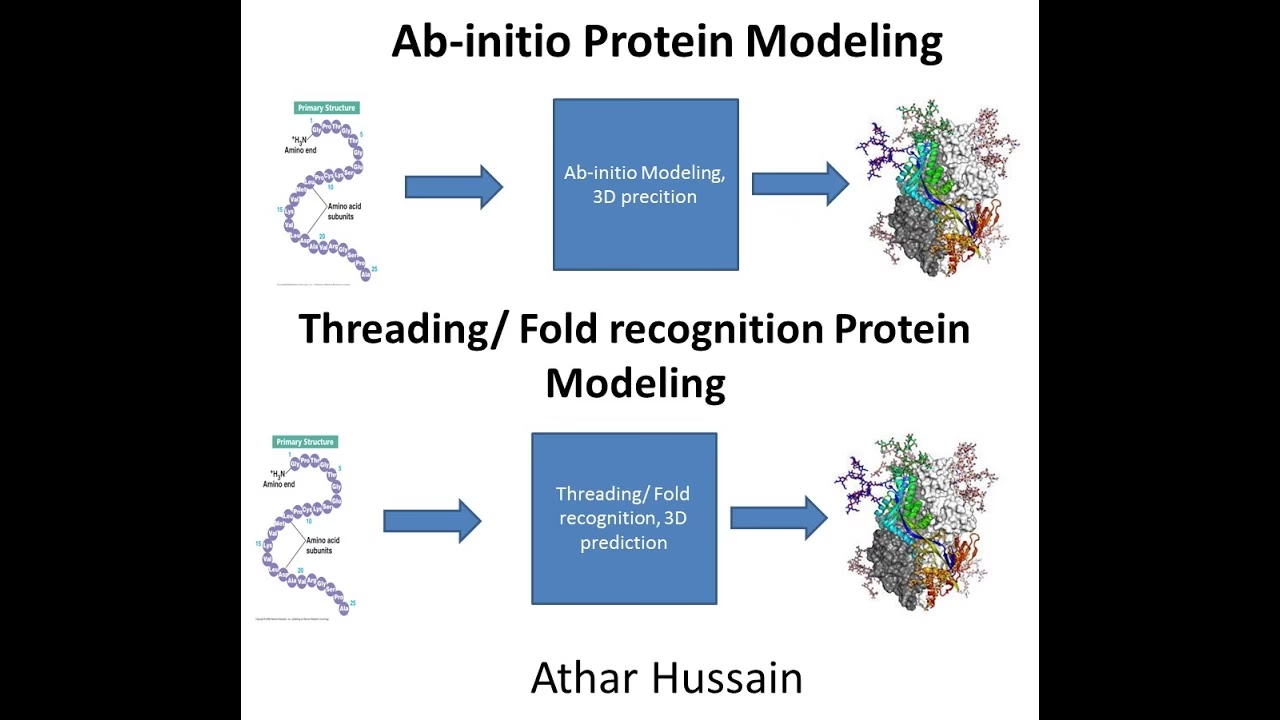
Mere teknisk sagt er proteintrådning en beregningsmetode, der bruges til at forudsige 3D-strukturen af et protein, hvis aminosyresekvens er kendt. Metoden fungerer ved at sammenligne målsekvensen med et bibliotek af kendte proteinstrukturer (kaldet 'folds'). Algoritmen vurderer, hvor godt aminosyrerne i målsekvensen passer ind i de forskellige strukturelle miljøer i hver kendt fold. Den fold, der giver det bedste 'match' eller den laveste 'energiscore', vælges som den mest sandsynlige struktur for det ukendte protein.
Processen Bag Proteintrådning: Et Trin-for-Trin Overblik
Selvom det lyder simpelt i teorien, er den faktiske proces en kompleks beregningsmæssig udfordring. Den kan generelt opdeles i følgende trin:
1. Oprettelse af et Fold-Bibliotek
Grundlaget for al proteintrådning er et omfattende bibliotek af kendte proteinstrukturer. Dette bibliotek, ofte kaldet et fold-bibliotek, er primært baseret på data fra Protein Data Bank (PDB). PDB er et globalt arkiv, der indeholder tusindvis af proteinstrukturer, som er blevet bestemt eksperimentelt gennem teknikker som røntgenkrystallografi og NMR-spektroskopi. Disse kendte strukturer fungerer som de skabeloner, man vil teste målsekvensen imod.
2. Definition af en Scorefunktion
Kernen i enhver trådningsmetode er en scorefunktion (også kaldet en potentiale- eller energifunktion). Denne funktion er designet til at vurdere, hvor 'glad' en bestemt aminosyre er for at befinde sig i et specifikt strukturelt miljø. Funktionen tager højde for flere faktorer, såsom:
- Parvise interaktioner: Hvordan interagerer aminosyrer, der er langt fra hinanden i sekvensen, men tæt på hinanden i den foldede struktur? Nogle par af aminosyrer tiltrækker hinanden, mens andre frastøder hinanden.
- Solvatisering: Passer det, at en hydrofob (vandskyende) aminosyre er begravet inde i proteinets kerne, væk fra vand, mens en hydrofil (vandelskende) aminosyre er på overfladen?
- Sekundær struktur: Passer sekvensen med de forudsagte sekundære strukturelementer (alfa-helixer og beta-strenge) i skabelonfolden?
Målet er at finde den justering af sekvensen på folden, der minimerer den samlede energi ifølge scorefunktionen.
3. Sekvens-Struktur Alignment
Dette er selve 'trådnings'-trinnet. En algoritme forsøger at placere hver aminosyre fra målsekvensen på en position i skabelonstrukturen. Dette er mere kompliceret end en simpel sekvens-til-sekvens alignment, fordi algoritmen skal tage højde for den tredimensionelle kontekst. Den skal kunne håndtere 'gaps', hvilket svarer til, at dele af perlesnoren (sekvensen) danner løkker, der stikker ud fra ståltrådsskulpturen (folden).
4. Vurdering og Udvælgelse
Efter at have trådet målsekvensen gennem mange forskellige folds i biblioteket og beregnet en score for hver, skal den bedste model identificeres. Den alignment, der resulterer i den mest favorable score (typisk den laveste energi), anses for at være den mest sandsynlige forudsigelse. Ofte bruges statistiske metoder til at vurdere, hvor signifikant en score er, for at skelne en ægte forudsigelse fra et tilfældigt match.
Sammenligning med Andre Metoder
Proteintrådning er en af tre hovedkategorier af metoder til forudsigelse af proteinstruktur. Det er nyttigt at forstå, hvordan de adskiller sig fra hinanden.
| Metode | Princip | Hvornår bruges den? | Forventet Nøjagtighed |
|---|---|---|---|
| Homologimodellering | Bruger en kendt struktur fra et meget nært beslægtet protein (en homolog) som skabelon. | Når der findes et protein med høj sekvenslighed (>30-40%) og kendt struktur. | Høj |
| Proteintrådning (Fold Recognition) | Matcher sekvensen mod et bibliotek af kendte folds, selv uden tydelig sekvenslighed. | Når der ikke findes en nær homolog, men proteinet forventes at have en allerede kendt fold. | Moderat til Høj |
| Ab Initio (De Novo) Forudsigelse | Forsøger at forudsige strukturen fra bunden, kun baseret på fysiske og kemiske principper, uden en skabelon. | Når proteinet har en helt ny fold, som ikke er set før. | Lav til Moderat |
Anvendelser og Betydning i Medicin og Forskning
Evnen til at forudsige en proteins 3D-struktur ud fra dens sekvens er en af de hellige graler inden for biologi. Det har enorme praktiske konsekvenser:
- Lægemiddeludvikling: Mange lægemidler virker ved at binde sig til et specifikt protein og enten hæmme eller aktivere det. Ved at kende proteinets 3D-struktur kan forskere designe lægemiddelmolekyler, der passer perfekt ind i proteinets aktive site, ligesom en nøgle i en lås. Dette gør udviklingen af nye lægemidler hurtigere og mere målrettet.
- Forståelse af Sygdomme: Mange genetiske sygdomme, som f.eks. cystisk fibrose og visse former for kræft, skyldes mutationer, der ændrer en enkelt aminosyre i et protein. Dette kan få proteinet til at folde forkert og miste sin funktion. Proteintrådning kan hjælpe med at forudsige, hvordan sådanne mutationer påvirker strukturen og dermed funktionen, hvilket giver indsigt i sygdommens molekylære årsag.
- Funktionel Genomik: Med moderne DNA-sekventeringsteknologier kan vi hurtigt bestemme alle generne i en organisme. Men en liste af gener er ikke nok – vi vil vide, hvad proteinerne, de koder for, gør. Strukturforudsigelse er et afgørende skridt i at tildele en funktion til et ukendt protein.
Udfordringer og Fremtiden
Selvom proteintrådning er en stærk metode, har den sine begrænsninger. Den største er, at den er afhængig af det eksisterende bibliotek af kendte folds. Hvis et protein har en helt ny og unik struktur, kan trådning ikke finde et match. Derudover er udviklingen af præcise scorefunktioner en vedvarende udfordring. At fange alle de subtile fysiske og kemiske interaktioner, der styrer proteinfoldning, i en simpel matematisk funktion er ekstremt svært.
Fremtiden ser dog lys ud. Med fremkomsten af kunstig intelligens og machine learning, især deep learning-metoder som dem, der bruges i AlphaFold, er nøjagtigheden af strukturforudsigelser steget dramatisk. Disse nye metoder lærer komplekse mønstre fra de enorme mængder af sekvens- og strukturdata, hvilket forbedrer alle aspekter af processen, fra alignment til scoring. Kombinationen af traditionelle trådningsprincipper med avanceret AI vil fortsat revolutionere vores evne til at afkode livets molekylære maskineri.
Ofte Stillede Spørgsmål (FAQ)
Hvad er forskellen på proteintrådning og homologimodellering?
Homologimodellering bruges, når dit målprotein har en meget høj grad af lighed i aminosyresekvensen med et protein med en kendt struktur. Det er som at bygge en model af en ny bil baseret på en næsten identisk tidligere model. Proteintrådning bruges, når der er meget lav eller ingen sekvenslighed, men man har en formodning om, at den overordnede fold kan være den samme. Det er som at erkende, at selvom to forskellige bilmærker ser forskellige ud, deler de det samme grundlæggende chassis.
Hvor nøjagtig er proteintrådning?
Nøjagtigheden kan variere meget. Hvis målproteinet har en fold, der er godt repræsenteret i biblioteket, kan forudsigelsen være meget god. Hvis matchet er fjernt, eller hvis proteinet har unikke løkker eller domæner, vil nøjagtigheden være lavere. Generelt er den mindre præcis end homologimodellering, men betydeligt bedre end ab initio-metoder for proteiner med kendte folds.
Hvad er en 'fold' i proteiners verden?
En 'fold' refererer til den overordnede arkitektur eller topologi af et protein. Proteiner med meget forskellige sekvenser og funktioner kan nogle gange folde sig til en meget lignende overordnet form. Der menes at være et begrænset antal (måske et par tusinde) grundlæggende folds i naturen, som genbruges igen og igen.
Hvis du vil læse andre artikler, der ligner Proteintrådning: Afkodning af Livets Byggesten, kan du besøge kategorien Sundhed.