29/04/2017
I en verden, hvor medicinsk teknologi udvikler sig med lynets hast, spiller medicinsk udstyr en stadig mere central rolle i vores sundhedssystem. Fra simple blodtryksmålere til avancerede pacemakere og kirurgiske robotter er vi afhængige af, at disse produkter er både sikre og effektive. Men hvem sikrer, at producenterne lever op til de højeste standarder? I USA er det primært Food and Drug Administration (FDA), der bærer dette tunge ansvar. FDA's rolle er dog ikke kun at agere som en streng kontrollant; de fungerer også som en afgørende partner og vejleder for industrien for at fremme innovation og sikre, at patienter får adgang til den bedst mulige teknologi på en sikker måde.
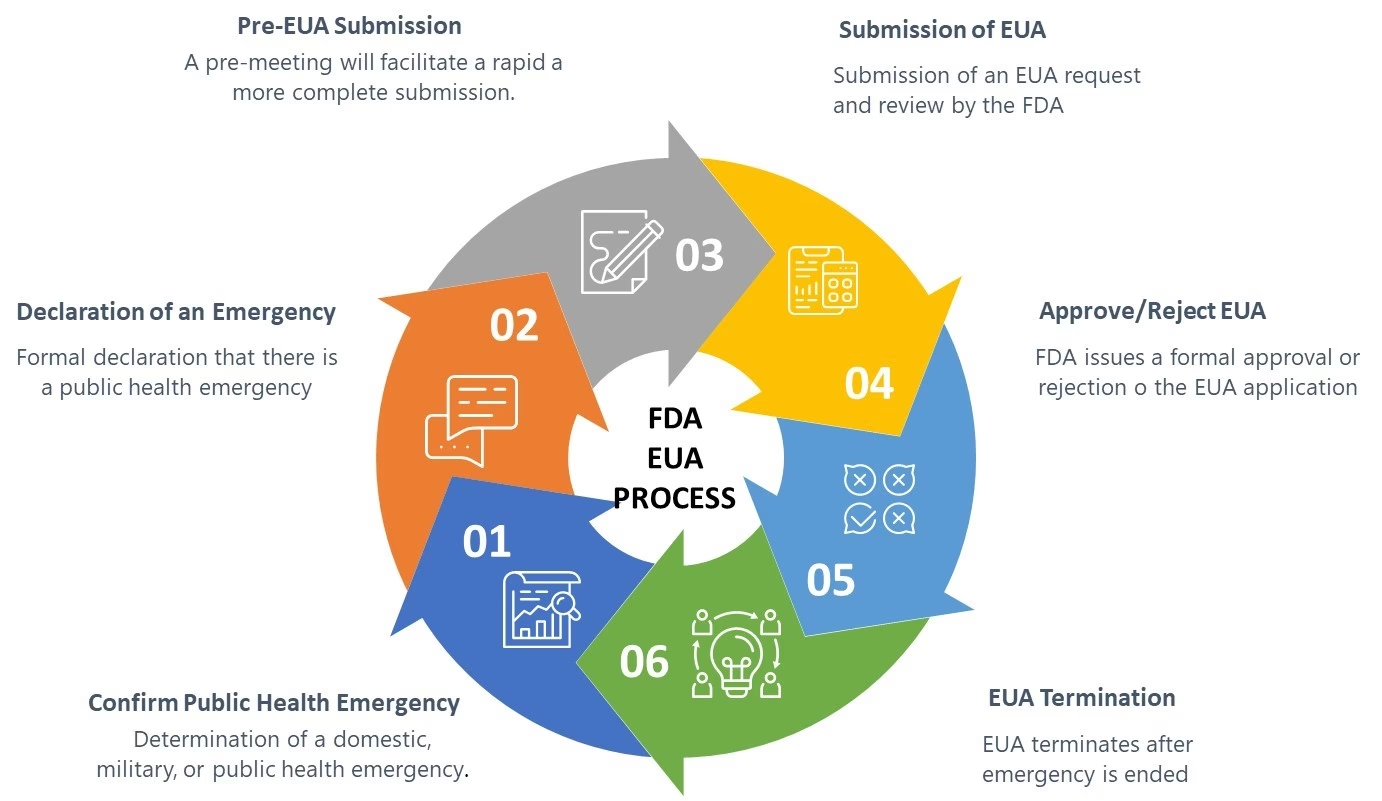
Hvad er FDA, og Hvorfor er de Vigtige?
FDA er en føderal myndighed under det amerikanske sundhedsministerium. Deres primære mission er at beskytte den offentlige sundhed ved at sikre sikkerheden, effektiviteten og sikringen af humane og veterinære lægemidler, biologiske produkter og medicinsk udstyr. De er også ansvarlige for at sikre sikkerheden af landets fødevareforsyning, kosmetik og produkter, der udsender stråling.
Når det kommer til medicinsk udstyr, er det FDA's Center for Devices and Radiological Health (CDRH), der tager føringen. Deres vision er, at patienter og sundhedspersonale skal have adgang til sikre, effektive og højkvalitets medicinske anordninger og strålingsudsendende produkter. For at opnå dette har FDA udviklet en række programmer og vejledninger, der hjælper industrien med at forstå og overholde de komplekse regulatoriske krav.
FDA's Vejledning til Industrien: Et Partnerskab for Sikkerhed
Man kunne forestille sig, at forholdet mellem en regulerende myndighed og industrien er præget af modstand. Men FDA har indset, at den bedste vej til patientsikkerhed er gennem klar kommunikation og samarbejde. Derfor udgiver de en lang række vejledningsdokumenter. Disse dokumenter er ikke love i sig selv, men de repræsenterer FDA's aktuelle tænkning om et givent emne. De giver industrien et klart billede af, hvad FDA forventer, og hvordan man bedst kan navigere i godkendelsesprocessen.
Disse vejledninger dækker et bredt spektrum af emner, herunder:
- Kliniske forsøgsdesign
- Krav til mærkning og markedsføring
- Cybersikkerhed for netværksforbundet udstyr
- Kvalitetssystemregulering (QSR)
- Og, meget centralt, procesvalidering
Ved at tilbyde denne vejledning hjælper FDA virksomheder med at undgå almindelige faldgruber, hvilket kan spare dem for både tid og penge. Målet er at skabe en forudsigelig og gennemsigtig proces, der tilskynder til udvikling af nyt og forbedret medicinsk udstyr, uden at gå på kompromis med patientens sikkerhed.
Kernen i Kvalitetssikring: FDA's Retningslinjer for Procesvalidering
Et af de mest kritiske områder, FDA fokuserer på, er procesvalidering. Men hvad betyder det egentlig? Kort sagt er procesvalidering den proces, hvor en producent indsamler og evaluerer data fra designstadiet og gennem hele produktionsprocessen for at etablere videnskabeligt bevis for, at processen er i stand til konsekvent at levere et kvalitetsprodukt.
Forestil dig en producent af kunstige hjerteklapper. Det er ikke nok, at de producerer én perfekt hjerteklap. De skal kunne bevise, at deres produktionsproces kan fremstille tusindvis af identiske hjerteklapper, der alle lever op til de nøjagtige specifikationer, gang på gang. Den mindste afvigelse kan have fatale konsekvenser. Her er validering nøglen.
FDA's vejledning om procesvalidering skitserer principper og praksisser, som agenturet anser for acceptable. Et vigtigt punkt i denne vejledning er, at den ikke opstiller en liste over rigide regler, der skal følges slavisk. I stedet anerkender den, at forskellige produkter og processer kan kræve forskellige tilgange. Denne fleksibilitet er afgørende. Den giver producenterne mulighed for at anvende de nyeste teknologier og metoder, så længe de kan bevise, at deres valgte metode er videnskabeligt forsvarlig og fører til et konsistent og sikkert produkt.
De Tre Stadier af Procesvalidering
FDA's moderne tilgang til procesvalidering er typisk opdelt i tre faser:
- Fase 1: Procesdesign (Process Design): Denne fase fokuserer på at designe processen. Her indsamles viden fra produktudvikling og eksperimenter for at forstå de potentielle kilder til variation og etablere en strategi for proceskontrol. Målet er at designe en proces, der er robust og pålidelig.
- Fase 2: Proceskvalificering (Process Qualification): I denne fase evalueres procesdesignet for at bekræfte, at det er i stand til at fungere reproducerbart i kommerciel skala. Dette indebærer en grundig test af anlægget, udstyret og personalets ydeevne. Man producerer typisk et antal test-batches for at demonstrere, at processen er under kontrol.
- Fase 3: Fortsat Procesverifikation (Continued Process Verification): Validering stopper ikke, når produktet rammer markedet. Denne fase kræver løbende overvågning under rutinemæssig produktion for at sikre, at processen forbliver i en valideret tilstand. Eventuelle uventede afvigelser skal undersøges, og der skal træffes korrigerende foranstaltninger.
Denne livscyklustilgang sikrer, at produktets kvalitet ikke kun er noget, der opnås i starten, men noget der vedligeholdes gennem hele produktets levetid.
Sammenligning: FDA's Vejledning vs. Strikte Regler
For at illustrere fordelene ved FDA's tilgang kan vi sammenligne den med en mere rigid, regelbaseret model.
| Karakteristik | FDA's Vejledningsmodel | Strikt Regelbaseret Model |
|---|---|---|
| Fleksibilitet | Høj. Tillader brug af nye teknologier og metoder, så længe de er videnskabeligt validerede. | Lav. Kræver, at alle følger de samme, prædefinerede trin, uanset produktets art. |
| Innovation | Fremmer innovation ved at give plads til nye løsninger og forbedringer. | Kan hæmme innovation, da det er svært at implementere nye metoder, der ikke passer ind i de faste regler. |
| Ansvar | Placerer et større ansvar hos producenten for at bevise, at deres proces er effektiv og sikker. | Fokuserer mere på, om producenten har fulgt reglerne, end på det endelige resultat. |
| Opdatering | Vejledninger kan opdateres relativt hurtigt for at afspejle ny videnskab og teknologi. | Lovgivning og faste regler er ofte langsomme og besværlige at ændre. |
Ofte Stillede Spørgsmål (FAQ)
Gælder FDA's regler også i Danmark?
Nej, ikke direkte. Danmark og resten af EU har deres egne omfattende regler for medicinsk udstyr, kendt som Forordningen om Medicinsk Udstyr (MDR). Produkter, der sælges i Europa, skal have et CE-mærke, som viser, at de overholder disse regler. Dog er FDA-godkendelse anerkendt som en af de højeste standarder globalt. Mange internationale virksomheder søger både CE-mærkning og FDA-godkendelse, og de to systemer deler mange grundlæggende principper om sikkerhed og effektivitet.
Er et FDA-godkendt produkt 100% risikofrit?
Intet medicinsk produkt er fuldstændig uden risiko. FDA's godkendelse betyder, at agenturet har fastslået, at fordelene ved produktet for den påtænkte patientgruppe opvejer de kendte risici. FDA har også et robust system til overvågning af produkter, efter de er kommet på markedet (post-market surveillance), for at opdage eventuelle nye eller uventede problemer.
Hvad sker der, hvis en producent ikke følger FDA's vejledninger?
Selvom vejledninger teknisk set ikke er lov, er det i praksis meget svært at få et produkt godkendt uden at følge dem eller kunne præsentere en alternativ, lige så robust tilgang. Hvis en producent ikke kan demonstrere, at deres processer er validerede og under kontrol, vil de ikke få markedsføringstilladelse. For produkter, der allerede er på markedet, kan manglende overholdelse føre til advarselsbreve, bøder, tilbagekaldelse af produkter og i værste fald retsforfølgelse.
Hvorfor er det vigtigt, at FDA's vejledninger kan ændres?
Medicinsk videnskab og teknologi er i konstant udvikling. En vejledning, der var passende for ti år siden, er måske ikke længere tilstrækkelig i dag. Ved at anerkende, at deres vejledninger kan og skal ændres over tid, sikrer FDA, at deres regulering forbliver relevant og i stand til at håndtere nye udfordringer, som f.eks. kunstig intelligens i diagnostisk udstyr eller avancerede nye materialer. Dette sikrer, at reguleringen både beskytter patienterne og understøtter fremskridt.
Hvis du vil læse andre artikler, der ligner Sådan Sikrer FDA Kvaliteten af Medicinsk Udstyr, kan du besøge kategorien Sundhed.