22/07/2015
Det europæiske marked for lægemidler er et af verdens største og mest attraktive, men adgang hertil er strengt reguleret for at sikre patienternes sikkerhed og produkternes kvalitet. For enhver producent, der ønsker at eksportere medicin til Den Europæiske Union (EU), er det afgørende at forstå og overholde de gældende regler. Kernen i denne regulering er den såkaldte fremstillings- og importtilladelse, bedre kendt som en MIA (Manufacturing and Import Authorization). Denne tilladelse er din nøgle til lovligt at kunne fremstille eller importere lægemidler inden for EU's grænser. Uden en gyldig MIA er det ulovligt at bringe lægemidler ind på det europæiske marked. Denne artikel vil guide dig igennem de væsentlige aspekter af MIA-processen, hvem der har brug for tilladelsen, og hvordan du som producent, både i og uden for EU, skal forholde dig.
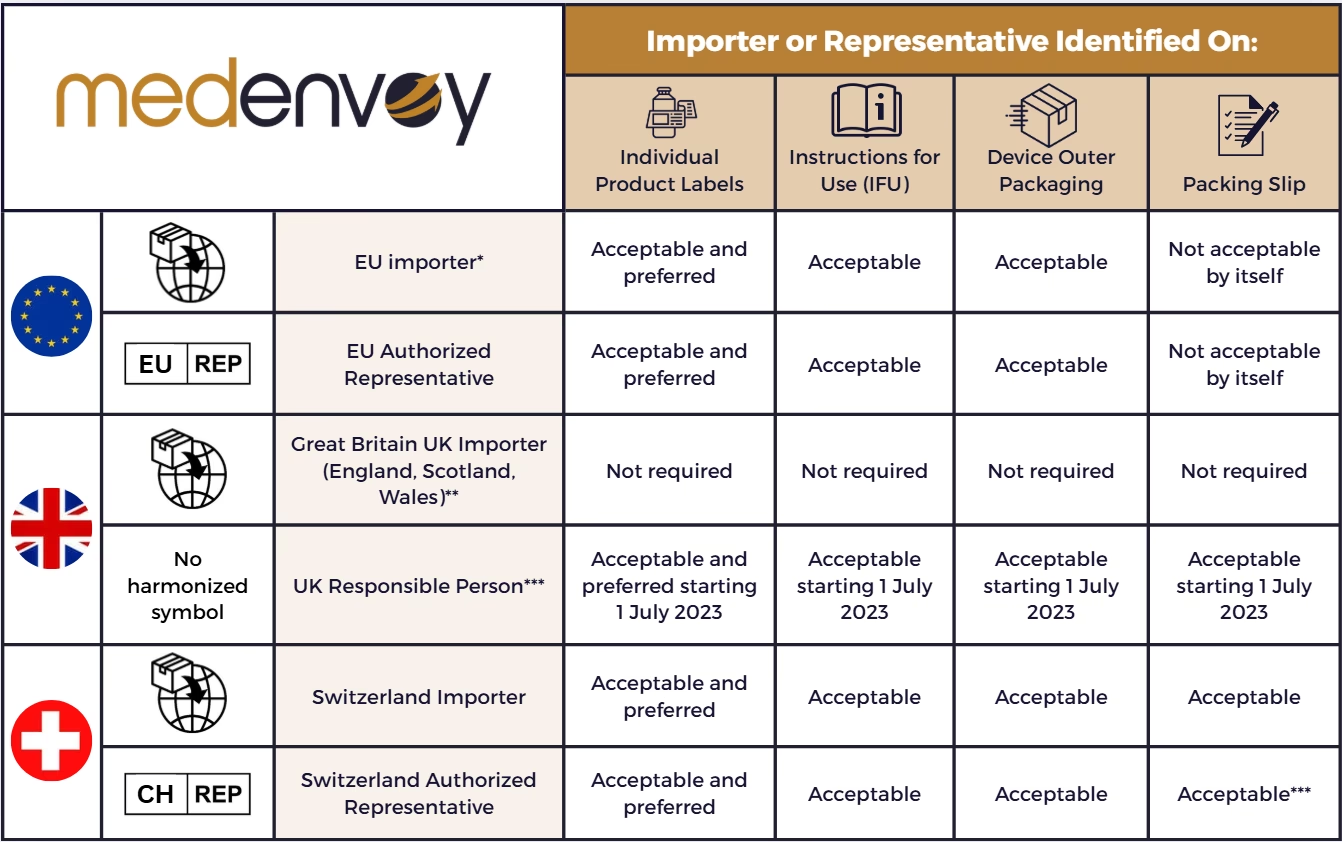
- Hvad er en fremstillings- og importtilladelse (MIA)?
- Hvem har brug for en MIA-tilladelse?
- Processen for at opnå en MIA i EU
- Særlige regler for producenter uden for EØS
- Forskel på aktive stoffer (API) og færdige produkter (FP)
- Rækkevidden af din godkendelse: Én importør vs. hele EU
- Nødvendig dokumentation for import
- Ofte Stillede Spørgsmål (FAQ)
Hvad er en fremstillings- og importtilladelse (MIA)?
En fremstillings- og importtilladelse (MIA) er en officiel licens udstedt af en national kompetent myndighed i et EU/EØS-land. Denne licens bekræfter, at en virksomhed overholder principperne for god fremstillingspraksis, eller GMP (Good Manufacturing Practice). Selvom det officielle navn er MIA, refererer mange i branchen til den som en "GMP-licens" eller "GMP-godkendelse", da overholdelse af GMP-reglerne er den absolutte forudsætning for at opnå og vedligeholde tilladelsen. GMP er et kvalitetssikringssystem, der sikrer, at lægemidler produceres og kontrolleres konsekvent efter de kvalitetsstandarder, der passer til deres tilsigtede anvendelse. Det dækker alle aspekter af produktionen, fra råmaterialer, lokaler og udstyr til personalets uddannelse og hygiejne.
Hvem har brug for en MIA-tilladelse?
Behovet for en MIA er meget specifikt og afhænger af din virksomheds rolle og placering. Du skal have en gyldig MIA, hvis din virksomhed opfylder en af de følgende to betingelser:
- Import fra lande uden for EU: Hvis du importerer GMP-regulerede lægemidler fra et land uden for Den Europæiske Union (et såkaldt tredjeland) med henblik på at markedsføre dem i EU. I dette tilfælde fungerer din virksomhed som den juridisk ansvarlige enhed, der sikrer, at de importerede produkter lever op til EU's standarder.
- Fremstilling inden for EU: Hvis du er en producent af lægemidler og har dine produktionsfaciliteter placeret inden for et EU-land. Dette gælder for alle former for fremstilling, herunder fuld produktion, delvis produktion, pakning og frigivelse af batches.
Det er vigtigt at bemærke, at en MIA er knyttet til en specifik juridisk enhed og en fysisk adresse. Den er ikke en generel godkendelse, der kan overføres frit.
Processen for at opnå en MIA i EU
At opnå en MIA er en formel og grundig proces, der ikke kan sammenlignes med at anmode om en standardcertificering som f.eks. ISO. Du kan ikke blot hyre et firma til at inspicere dig og derefter udstede et certifikat. Processen involverer de nationale lægemiddelmyndigheder.
Trin 1: Ansøgning hos den nationale myndighed
Først skal du registrere din juridiske enhed hos den relevante nationale reguleringsmyndighed i det EU-land, hvor din virksomhed er baseret. I Danmark er den kompetente myndighed Lægemiddelstyrelsen. I Holland er det Farmatec, og i Tyskland er det Paul-Ehrlich-Institut eller BfArM, afhængigt af produkttypen. Hvert land har sin egen myndighed, og en komplet liste kan findes på hjemmesiden for Det Europæiske Lægemiddelagentur (EMA).
Trin 2: Planlægning af inspektion
Når din ansøgning er modtaget og accepteret, vil myndigheden planlægge en inspektion af dine faciliteter. Myndighederne anvender en risikobaseret tilgang til planlægning. Det betyder, at anlæg, der anses for at være kritiske – f.eks. producenter af vacciner under en pandemi eller producenter af sterile produkter – vil blive prioriteret højere end anlæg, der producerer mindre kritiske produkter. Ventetiden kan derfor variere betydeligt.
Trin 3: Forberedelse til inspektion
Dette er den mest kritiske fase. Du skal sikre dig, at din virksomhed er fuldt forberedt til den regulatoriske inspektion. Det indebærer, at du skal have implementeret alle krav i EU's GMP-regelsæt samt eventuelle yderligere lokale lovkrav. Din forberedelse bør omfatte en grundig gennemgang af:
- Dit kvalitetssikringssystem
- Dokumentationspraksis (SOP'er, batch-journaler, valideringsrapporter)
- Personalets kvalifikationer og træning
- Lokalernes og udstyrets egnethed og vedligeholdelse
- Råvarekontrol og leverandørstyring
- Produktions- og proceskontrol
- Kvalitetskontrol og testning
- Håndtering af afvigelser, klager og tilbagekaldelser
En mangelfuld forberedelse kan føre til afvisning af din ansøgning, hvilket kan forårsage betydelige forsinkelser og omkostninger.
Særlige regler for producenter uden for EØS
For virksomheder, der er placeret uden for Det Europæiske Økonomiske Samarbejdsområde (EØS), er processen anderledes. En producent i f.eks. USA, Indien eller Kina kan ikke selv opnå en MIA-tilladelse fra en EU-myndighed.
Ansvaret ligger i stedet hos den europæiske importør. Det er importøren, der er baseret i EØS, som skal have en MIA for at kunne importere dine produkter. Som en del af importørens ansvar skal denne sikre, at du som producent i tredjelandet overholder standarder, der er mindst ækvivalente med EU-GMP. Dette indebærer typisk, at importøren udfører en grundig audit af dine produktionsfaciliteter, før de kan begynde at importere dine produkter. Importøren bliver dermed den juridiske garant over for EU-myndighederne for, at de importerede lægemidler er fremstillet korrekt.
Forskel på aktive stoffer (API) og færdige produkter (FP)
Kravene til dig som producent uden for EØS afhænger af, hvilken type produkt du eksporterer. Der er en væsentlig forskel på, om du fremstiller aktive farmaceutiske ingredienser (API'er) eller færdige lægemidler (Finished Products - FP), der er klar til patienten.
I tilfælde af Aktive Farmaceutiske Ingredienser (API'er):
Hvis du kun producerer den aktive substans, som senere skal formuleres til et færdigt lægemiddel af en producent i EU, er kravene lidt lempeligere. Hvis du består din europæiske importørs GMP-audit, bliver du betragtet som "GMP-certificeret via proxy". Det betyder, at importørens godkendelse er tilstrækkelig dokumentation. Du vil sandsynligvis ikke blive inspiceret direkte af en EU-myndighed, men du vil alligevel kunne eksportere dine API'er til EU via denne importør.
I tilfælde af Færdige Produkter (FP):
Hvis du eksporterer færdige produkter – altså medicin, der ikke skal bearbejdes yderligere og går direkte til patienten – er kravene strengere. Her er det ikke nok med en audit fra importøren. Du skal have et officielt EU GMP-certifikat. Dette certifikat kan kun udstedes af en kompetent EU-myndighed efter en vellykket inspektion af dine faciliteter. Dette er en meget mere omfattende proces, der kræver direkte interaktion med de europæiske myndigheder.
Sammenligning af krav for API og FP
| Kriterium | Aktive Farmaceutiske Ingredienser (API) | Færdige Lægemidler (FP) |
|---|---|---|
| Ansvarlig for GMP-kontrol | EU-importøren via audit | EU-myndighed via direkte inspektion |
| Nødvendig godkendelse | Bestået audit fra importør ("certificeret via proxy") | Officielt EU GMP-certifikat |
| Adgang til EU-markedet | Kun via den specifikke importør, der har auditeret | Adgang for alle importører i hele EU |
Rækkevidden af din godkendelse: Én importør vs. hele EU
Det er afgørende at forstå begrænsningerne ved en godkendelse, der udelukkende er baseret på en importørs audit. Selvom din importør certificerer dig som værende i overensstemmelse med EU-GMP, gælder denne godkendelse kun for forretningsforholdet med netop denne importør. Hvis du ønsker at sælge dine produkter til en anden importør i et andet EU-land, skal denne nye importør udføre sin egen, separate audit af dine faciliteter. Hver importør er juridisk forpligtet til selvstændigt at verificere sine leverandører.
Den eneste måde at opnå bred adgang til hele EU-markedet på er ved at blive inspiceret af en EU-reguleringsmyndighed og modtage et officielt GMP-certifikat. Når du har dette certifikat, er det registreret i EudraGMDP-databasen og anerkendes af alle nationale myndigheder og importører i hele EU. Dette forenkler processen markant, hvis du planlægger at arbejde med flere partnere i Europa.
Nødvendig dokumentation for import
Udover GMP-overholdelse kræver importprocessen specifik dokumentation. For hvert lægemiddel, der importeres, skal der indsendes en kopi af producentens fremstillingstilladelse. Det er vigtigt at bemærke, at dette skal inkludere de relevante bilag, der specificerer produktionsstedet og de tilladte fremstillingsaktiviteter. Det er typisk ikke nødvendigt at fremlægge detaljerede plantegninger. Hvis de originale tilladelser ikke er på tysk eller engelsk, skal der vedlægges en autoriseret oversættelse til et af disse sprog.
Ofte Stillede Spørgsmål (FAQ)
Kan min virksomhed uden for EU ansøge direkte om en MIA?
Nej, en virksomhed uden for EØS kan ikke selv ansøge om eller opnå en MIA. Det er din partner – importøren, som er baseret i EU/EØS – der skal have en MIA for at importere dine produkter. Ansvaret for at verificere din GMP-overholdelse ligger hos dem.
Hvad er forskellen på en MIA og en GMP-licens?
I praksis er der ingen forskel. MIA (Manufacturing and Import Authorization) er det officielle juridiske navn for tilladelsen. GMP-licens er et mere almindeligt anvendt udtryk i branchen, da overholdelse af GMP er forudsætningen for at få tilladelsen.
Er en godkendelse fra én importør nok til at sælge i hele EU?
Nej. En audit og godkendelse fra en enkelt importør kvalificerer dig kun til at levere til den specifikke importør. Hver ny importør skal udføre sin egen uafhængige audit. For at få adgang til at levere til alle importører i EU kræves et officielt GMP-certifikat udstedt af en EU-myndighed.
Hvilken myndighed skal jeg kontakte i Danmark?
I Danmark er den kompetente myndighed, der udsteder MIA-tilladelser og udfører GMP-inspektioner, Lægemiddelstyrelsen. Det er dem, en dansk-baseret importør eller producent skal ansøge hos.
At navigere i EU's regulatoriske landskab kan virke overvældende, men med grundig forberedelse og de rette partnere er det en overkommelig proces. Nøglen til succes er en dybdegående forståelse af GMP-kravene og et tæt samarbejde med din europæiske importør, som vil være din guide og juridiske repræsentant på det europæiske marked.
Hvis du vil læse andre artikler, der ligner Eksporter medicin til EU: Guide til MIA, kan du besøge kategorien Sundhed.