30/09/2024
Hyperparathyroidisme-Kæbetumor (HPT-JT) syndrom er en sjælden og kompleks arvelig lidelse, der ofte forbliver udiagnosticeret i lang tid på grund af dens varierende og til tider uspecifikke symptomer. Patienter kan præsentere sig med alt fra nyresten til en smertefri hævelse i kæben, hvilket gør det til en diagnostisk udfordring for læger. Sygdommen er kendetegnet ved en kombination af overaktive biskjoldbruskkirtler, godartede tumorer i kæben og en øget risiko for andre tumorer, især i nyrerne og livmoderen. At forstå denne tilstand er afgørende, da tidlig diagnose og korrekt håndtering markant kan forbedre en patients prognose og livskvalitet, især i betragtning af den forhøjede risiko for, at nogle af disse tumorer udvikler sig til kræft.
Hvad er Hyperparathyroidisme-Kæbetumor (HPT-JT) Syndrom?
Hyperparathyroidisme-Kæbetumor (HPT-JT) syndrom er en autosomal dominant arvelig sygdom. Det betyder, at hvis en forælder har den genetiske mutation, er der 50% chance for, at hvert barn arver den. Syndromet skyldes en mutation i CDC73-genet, som normalt fungerer som et tumorsuppressorgen, der hjælper med at kontrollere cellevækst og -deling. Når dette gen er muteret, mister det sin evne til at undertrykke tumordannelse, hvilket fører til de forskellige manifestationer af syndromet.
En af de mest bemærkelsesværdige egenskaber ved HPT-JT er dens variable ekspression og ufuldstændige penetrans. Dette betyder, at ikke alle, der arver genmutationen, vil udvikle alle symptomerne, og sværhedsgraden kan variere betydeligt fra person til person, selv inden for den samme familie. Nogle kan have alvorlig sygdom i en ung alder, mens andre kan have milde eller ingen symptomer hele livet. Denne uforudsigelighed gør genetisk rådgivning og regelmæssig screening for familiemedlemmer yderst vigtigt.
De primære kliniske træk ved HPT-JT syndrom omfatter:
- Primær hyperparathyroidisme (PHPT): Den mest almindelige manifestation, forårsaget af godartede tumorer (adenomer) i en eller flere af de fire biskjoldbruskkirtler.
- Ossificerende fibromer i kæben: Godartede, men lokalt aggressive tumorer, der udvikler sig i over- eller underkæben.
- Nyreproblemer: Kan omfatte cyster, godartede tumorer (hamartomer) og en øget risiko for nyrekræft (Wilms' tumor eller adenocarcinom).
- Livmodertumorer: Kvinder med HPT-JT har en øget risiko for at udvikle tumorer i livmoderen.
De Vigtigste Symptomer og Manifestationer
Symptomerne på HPT-JT er direkte relateret til de organer, der er påvirket. Ofte opdages de forskellige komponenter af syndromet på forskellige tidspunkter i en patients liv, hvilket kan forsinke den samlede diagnose.
Primær Hyperparathyroidisme (PHPT)
Dette er ofte det første og mest almindelige tegn på HPT-JT. Biskjoldbruskkirtlerne, som er små kirtler placeret bag skjoldbruskkirtlen i nakken, producerer parathyroideahormon (PTH), der regulerer kalciumniveauet i blodet. I HPT-JT udvikler en eller flere af disse kirtler et adenom, en godartet tumor, der producerer for meget PTH. Dette fører til primær hyperparathyroidisme, hvilket resulterer i et forhøjet kalciumniveau i blodet (hypercalcæmi).
Symptomerne på højt kalcium kan være vage og omfatter:
- Træthed og svaghed
- Øget tørst (polydipsi) og hyppig vandladning (polyuri)
- Forstoppelse
- Kvalme og nedsat appetit
- Nyresten (nefrolithiasis)
- Knogle- og ledsmerter
Det er vigtigt at bemærke, at patienter med HPT-JT har en markant højere risiko (omkring 15-24%) for, at deres parathyroidea-adenom udvikler sig til parathyroideakarcinom, en sjælden form for kræft, sammenlignet med den generelle befolkning med PHPT.
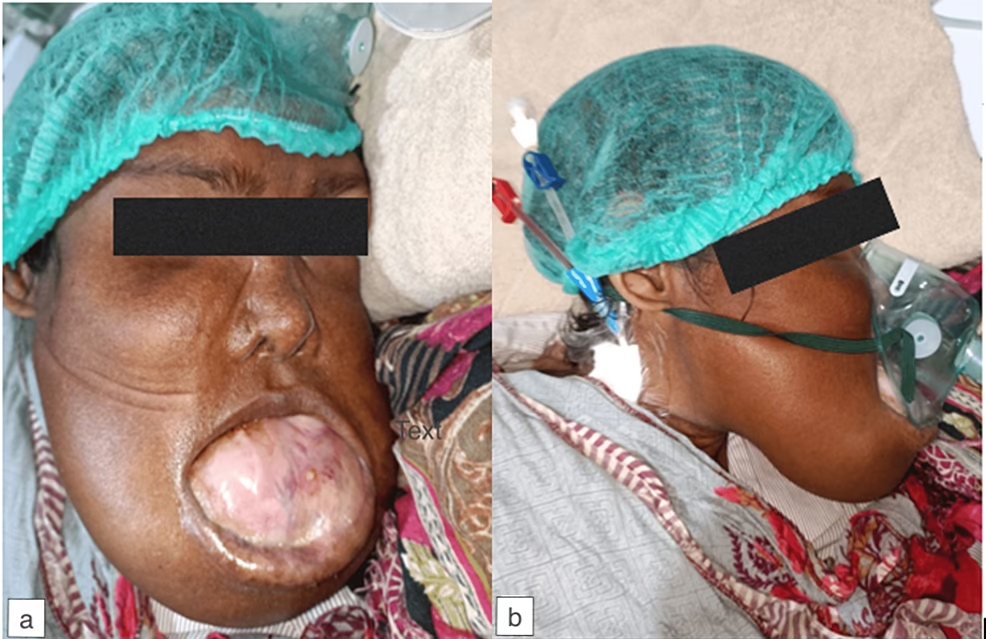
Ossificerende Fibromer i Kæben
Disse godartede, men potentielt vansirende tumorer er et kendetegn ved HPT-JT og findes hos omkring 30-50% af patienterne. Et ossificerende fibrom er en langsomt voksende tumor, der består af fibrøst væv og knoglelignende materiale. De opstår typisk i underkæben (mandibula) eller overkæben (maxilla) hos unge voksne eller teenagere.
Selvom de er godartede (risikoen for malign transformation er mindre end 0,5%), kan de være lokalt invasive. De kan:
- Forårsage en synlig, smertefri hævelse og ansigtsasymmetri.
- Forskyde tænderne.
- Vokse sig meget store og kompromittere luftvejene eller synet, hvis de strækker sig ind i bihulerne eller øjenhulen.
- Kræve omfattende kirurgi for fjernelse og efterfølgende rekonstruktion af kæben.
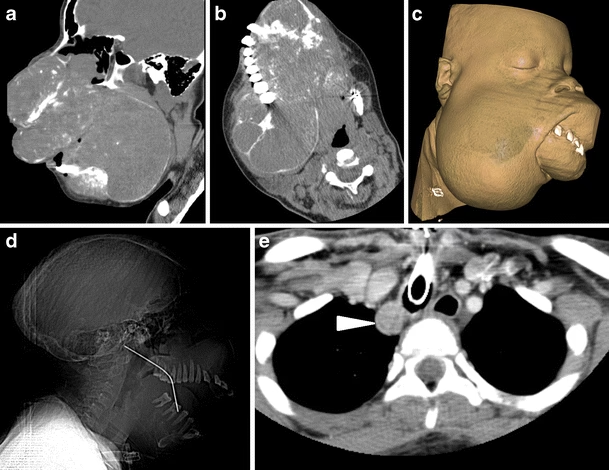
Diagnosen stilles ofte ved hjælp af røntgenbilleder (OPG) og CT-scanninger, hvor tumorerne fremstår som velafgrænsede, ekspansive læsioner. Deres udseende kan variere fra helt gennemsigtigt (radiolucent) til mere tæt (radiopaque), afhængigt af mængden af forkalket materiale i tumoren.
Diagnose og Udfordringer
At stille diagnosen HPT-JT kræver en høj grad af mistanke og en tværfaglig tilgang. En patient kan først henvende sig til en tandlæge for en kæbehævelse, en nefrolog for nyresten, eller en endokrinolog for symptomer på højt kalcium. Det er sammenhængen mellem disse tilsyneladende uafhængige problemer, der peger i retning af syndromet.
Diagnostiske skridt omfatter:
- Blodprøver: Måling af serumkalcium, fosfat og parathyroideahormon (PTH). Forhøjet kalcium og PTH med lavt fosfat er klassiske tegn på primær hyperparathyroidisme.
- Billeddiagnostik: Ultralyd af halsen og/eller en parathyroidea-scintigrafi bruges til at lokalisere det overaktive parathyroidea-adenom. Røntgen og CT-scanning af kæberne er afgørende for at identificere og vurdere ossificerende fibromer.
- Familiehistorie: En grundig familiehistorie er essentiel, da sygdommen er arvelig. Tilfælde af nyresten, kæbetumorer eller parathyroideasygdom hos slægtninge bør give anledning til mistanke.
- Genetisk testning: Den endelige bekræftelse af diagnosen sker ved en genetisk test, der påviser en mutation i CDC73-genet. Dette er ikke kun vigtigt for patienten, men også for at kunne tilbyde screening og rådgivning til familiemedlemmer.
Sammenligning af Kæbelæsioner
Det er vigtigt at skelne ossificerende fibromer fra andre kæbelæsioner. Her er en simpel sammenligning:
| Tilstand | Karakteristika | Røntgenudseende |
|---|---|---|
| Ossificerende Fibrom (HPT-JT) | Godartet, ekspansiv, velafgrænset tumor. | Velafgrænset, ofte med en tynd, sklerotisk kant. Kan være radiolucent, blandet eller radiopaque. |
| Fibrøs Dysplasi | Udviklingsforstyrrelse hvor normal knogle erstattes af fibrøst væv. | Diffus, dårligt afgrænset, "matteret glas"-udseende. |
| Brun Tumor | En knoglelæsion forårsaget af svær, ubehandlet hyperparathyroidisme. Ikke en ægte tumor. | Rent lytisk (knoglenedbrydende) læsion uden en sklerotisk kant. |
Behandling og Langsigtet Håndtering
Behandlingen af HPT-JT er rettet mod de specifikke manifestationer og kræver et team af specialister, herunder endokrinologer, kæbekirurger, nefrologer og kliniske genetikere.
- Behandling af Hyperparathyroidisme: Den primære behandling er kirurgisk fjernelse af det eller de forstørrede biskjoldbruskkirtler, en procedure kaldet parathyroidektomi. Dette normaliserer kalciumniveauerne og reducerer risikoen for komplikationer som nyresten og knogleskørhed.
- Behandling af Kæbetumorer: Ossificerende fibromer behandles med kirurgisk fjernelse. Afhængigt af tumorens størrelse og placering kan dette være en simpel udskrabning (enukleation) eller en mere omfattende resektion, der kan kræve rekonstruktion af kæben med knogletransplantater.
- Overvågning og Screening: På grund af den livslange risiko for nye tumorer og malign transformation er regelmæssig opfølgning afgørende. Dette inkluderer årlige blodprøver for kalcium og PTH, periodisk billeddiagnostik af kæberne og screening for nyre- og livmodertumorer. Familiemedlemmer, der er i risiko, bør også tilbydes genetisk testning og screening.
Ofte Stillede Spørgsmål (FAQ)
Er HPT-JT syndrom en form for kræft?
Ikke i sig selv. De fleste tumorer, der er forbundet med HPT-JT, såsom parathyroidea-adenomer og ossificerende fibromer i kæben, er godartede. Dog er der en markant forhøjet risiko for, at et parathyroidea-adenom udvikler sig til parathyroideakarcinom (kræft). Derfor er tidlig diagnose og behandling så vigtig.
Hvor almindelig er denne sygdom?
HPT-JT syndrom er meget sjældent. Den præcise forekomst er ukendt, men det anslås, at kun få hundrede familier på verdensplan er blevet diagnosticeret. Den sande forekomst kan dog være højere på grund af underdiagnosticering.
Hvis min forælder har HPT-JT, vil jeg så også få det?
Da det er en autosomal dominant arvelig lidelse, er der 50% chance for at arve genmutationen fra en berørt forælder. Det er dog ikke sikkert, at du vil udvikle de samme symptomer eller have samme sværhedsgrad. Genetisk rådgivning og testning anbefales for at afklare din risiko.
Kan man leve et normalt liv med HPT-JT?
Ja. Med en korrekt diagnose, en proaktiv behandlingsplan og livslang opfølgning kan de fleste patienter med HPT-JT håndtere deres tilstand effektivt og leve et fuldt og aktivt liv. Nøglen er at være opmærksom på symptomer og følge de anbefalede screeningsprotokoller for at fange eventuelle problemer tidligt.
Hvis du vil læse andre artikler, der ligner Forstå HPT-JT: En Sjælden Genetisk Sygdom, kan du besøge kategorien Sygdomme.