12/12/2010
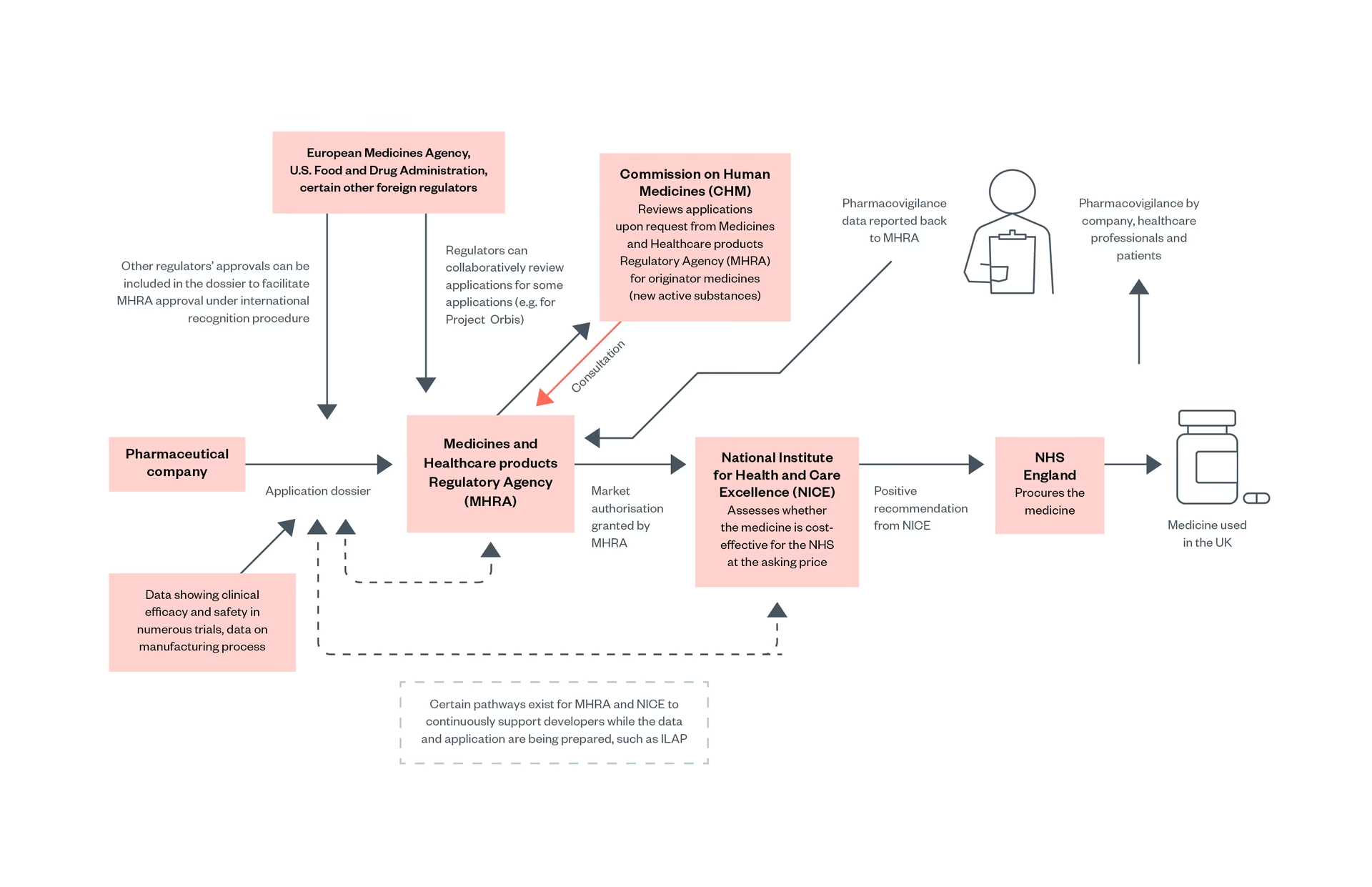
Den komplekse rejse mod et sikkert marked for medicinsk udstyr
I dag tager de fleste af os det for givet, at medicinsk udstyr som pacemakere, kunstige hofter og endda simple termometre er sikre at bruge. CE-mærket på emballagen fungerer som et stempel på sikkerhed og kvalitet. Men vejen til denne standardiserede regulering i Europa har været lang, kompleks og formet af både tragiske patientskandaler og stærke økonomiske interesser. Det var ikke en enkelt begivenhed, men en sammenløb af kriser, politisk vilje og industriens proaktive handlinger, der førte til det system, vi kender i dag. Denne artikel udforsker den fascinerende historie om, hvordan reguleringen af medicinsk udstyr i EU blev til, fra et kaotisk og fragmenteret landskab til de første skelsættende direktiver.
De tidlige dage: Et fragmenteret landskab
Før 1990'erne var Europa et kludetæppe af forskellige nationale regler for medicinsk udstyr. Systematisk regulering var stort set fraværende indtil midten af det 20. århundrede, simpelthen fordi teknologien endnu ikke var så avanceret. Tidlige forsøg på implantater, som Themistocles Glücks elfenbensledprotese i 1890, mislykkedes ofte. Først med teknologiske fremskridt som den første succesfulde implantation af en intraokulær linse i 1949 og den første fuldt implanterbare pacemaker i 1958, blev behovet for regulering tydeligt.
I 1990, da det første EU-direktiv blev vedtaget, var situationen i de 12 medlemslande meget forskelligartet. Nogle lande, som Italien, havde haft specifik lovgivning siden 1927, mens andre manglede dedikerede love. I fraværet af specifikke regler for udstyr, havde lande som Frankrig, Tyskland, Italien og Spanien tilpasset deres lægemiddellovgivning til også at dække visse typer udstyr. Tyskland skelnede mellem 'ægte lægemidler' og 'fiktive lægemidler' for at inkludere udstyr. Frankrig havde en godkendelsesprocedure kaldet 'homologation', der inkluderede inspektioner. I Storbritannien introducerede man procedurer for at registrere producenter og undersøge rapporter om defekter. Dette fragmenterede system skabte ikke kun potentielle sikkerhedsrisici for patienter, men også betydelige handelsbarrierer, der var i modstrid med selve grundlaget for Det Europæiske Økonomiske Fællesskab (EØF) – et fælles marked.
Vækkelseskaldene: Thalidomid og pacemaker-krisen
To store kriser, selvom den ene handlede om lægemidler, var afgørende for at skabe den politiske og offentlige vilje til strengere regulering.
Thalidomid-skandalen
Selvom Thalidomid var et lægemiddel og ikke medicinsk udstyr, blev skandalen en afgørende katalysator for al sundhedsregulering. Lægemidlet blev udviklet i 1953 og markedsført som et sikkert sovemiddel og et middel mod kvalme hos gravide kvinder. I Tyskland kunne det endda købes i håndkøb. Konsekvenserne var katastrofale: anslået 10.000 børn blev født med alvorlige misdannelser, især phocomelia (sællignende lemmer), og tusinder døde. Tragedien afslørede en chokerende mangel på krav til sikkerheds- og effektstudier før markedsføring. I Storbritannien, for eksempel, var der før Thalidomid ingen krav om bevis for klinisk sikkerhed eller effekt. Skandalen cementerede ideen om, at en kyndig og betroet tredjepart – en regulatorisk myndighed – er nødvendig for at beskytte patienternes interesser og afgøre, hvilke produkter der er sikre nok til at komme på markedet. Denne tankegang smittede af på debatten om medicinsk udstyr.
Pacemaker-problemerne
En mere direkte drivkraft for regulering af udstyr kom fra problemer med hjertepacemakere. Allerede i 1960'erne var der bekymringer, men i 1970'erne eskalerede det. I 1975 og 1976 måtte den store producent Medtronic tilbagekalde deres Xytron-pacemakere på grund af fugtindtrængning og batterisvigt, hvilket førte til en fejlrate på op til 1% om måneden og en betydelig risiko for pludselig død for patienterne. Tilbagekaldelsen påvirkede over 40.000 enheder. Da Medtronics europæiske juridiske direktør, Alan Howard, kontaktede sundhedsministerier i flere europæiske lande, opdagede han, at de fleste var uforberedte og usikre på, hvordan de skulle håndtere en tilbagekaldelse af udstyr. Der var simpelthen ingen lovgivning at læne sig op ad.
Denne krise fik producenterne selv til at handle. De indså, at et ureguleret marked var skadeligt for både patienter og forretning. I 1978 stiftede de International Association of Pacemaker Manufacturers (IAPM) for at kunne tale med én stemme og samarbejde med myndigheder om at etablere standarder og regler. Industrien var altså ikke en modstander, men en aktiv medspiller i skabelsen af regulering.
Det Indre Marked og 'Den Nye Tilgang'
Parallelt med disse sundhedskriser var en stærk økonomisk dagsorden i gang. Målet var at fuldføre EU's indre marked inden 1992, hvilket krævede fjernelse af tekniske handelsbarrierer. En skelsættende dom fra EF-Domstolen i 1979, 'Cassis de Dijon'-sagen, fastlagde princippet om gensidig anerkendelse: et produkt, der lovligt markedsføres i ét medlemsland, kan ikke forbydes i et andet. Dette var dog ikke nok.
I 1985 vedtog Rådet en resolution om 'Den Nye Tilgang' (New Approach) til teknisk harmonisering. Ideen var revolutionerende: i stedet for at EU-lovgivningen skulle specificere alle tekniske detaljer for et produkt, skulle den blot fastsætte de overordnede, essentielle krav til sikkerhed og ydeevne. Producenterne kunne derefter bruge harmoniserede europæiske standarder (udviklet af standardiseringsorganer som CEN og CENELEC) til at demonstrere, at de opfyldte disse krav. Overensstemmelsen skulle verificeres af uafhængige tredjeparter, de såkaldte Bemyndigede organer (Notified Bodies). Når et produkt var godkendt, kunne producenten påføre det et CE-mærke og frit sælge det i hele EØF. Denne model, drevet af et ønske om at styrke det indre marked, blev den skabelon, der skulle bruges for medicinsk udstyr.
Fødslen af den Første Lovgivning
I midten af 1980'erne henvendte både pacemaker-industrien (IAPM) og industrien for radiologisk udstyr (COCIR) sig uafhængigt af hinanden til Europa-Kommissionens Generaldirektorat for det Indre Marked (DG III). Industrien forklarede situationen og behovet for en europæisk løsning. Kommissionen, der på det tidspunkt manglede ekspertise inden for medicinsk udstyr, lyttede.
I 1987 blev en ekspertgruppe nedsat med repræsentanter fra nationale ministerier, industrien og klinikere for at hjælpe Kommissionen med at udarbejde et udkast til den første lovgivning. Det blev besluttet at starte med en fokuseret tilgang: et direktiv for aktive implanterbare medicinske anordninger. Dette var et logisk valg, da det dækkede de problematiske pacemakere og var en afgrænset, højrisikogruppe af produkter.
Industriorganisationerne spillede en afgørende rolle ved at levere forslag og teknisk ekspertise. Efter intense diskussioner i Rådet, hvor nogle lande som Tyskland argumenterede for et system mere lig det for lægemidler med krav om kliniske forsøg, blev man enige om at følge 'Den Nye Tilgang'. Den 20. juni 1990 blev en milepæl nået: Rådet vedtog Direktiv 90/385/EØF om aktivt, implantabelt medicinsk udstyr. Dette var den første bindende, fælleseuropæiske lovgivning for medicinsk udstyr og fundamentet for det system, der senere blev udvidet til at dække alt udstyr.
Tabel: Regulering Før og Efter 1990-direktivet
| Egenskab | Før 1990 | Efter Direktiv 90/385/EØF (for aktive implantater) |
|---|---|---|
| System | Fragmenteret; baseret på forskellige nationale love (eller mangel på samme). | Harmoniseret; baseret på fælles EU-regler. |
| Godkendelse | Varierede fra ingen krav til nationale godkendelser (f.eks. 'homologation' i Frankrig). | Overensstemmelsesvurdering af et Bemyndiget organ er påkrævet. |
| Markedsadgang | Produkt skulle godkendes i hvert land, hvilket skabte handelsbarrierer. | CE-mærket giver adgang til hele EU-markedet. |
| Krav til Producent | Uklare og inkonsistente krav på tværs af landegrænser. | Skal opfylde 'essentielle krav' til sikkerhed og ydeevne og have et kvalitetssystem. |
| Tilsyn | Begrænset og kun på nationalt niveau. Tilbagekaldelser var svære at koordinere. | Systematisk overvågning efter markedsføring og krav om indberetning af hændelser. |
Ofte Stillede Spørgsmål
Hvad var den direkte årsag til den første EU-lov om medicinsk udstyr?
Det var en kombination af faktorer. De mest direkte årsager var alvorlige sikkerhedsproblemer med højrisikoudstyr som pacemakere i 1970'erne, det stærke politiske og økonomiske ønske om at fjerne handelsbarrierer for at fuldføre EU's indre marked, og et proaktivt engagement fra industrien selv, som så fordelene i et forudsigeligt og harmoniseret regelsæt.
Hvorfor blev Thalidomid-skandalen vigtig for medicinsk udstyr, når det var en lægemiddelsag?
Thalidomid-skandalen skabte et fundamentalt skift i offentlighedens og politikernes opfattelse af produktsikkerhed. Den beviste, at det var bydende nødvendigt med en stærk, uafhængig reguleringsmyndighed til at beskytte patienter mod usikre produkter, før de kommer på markedet. Denne grundlæggende tankegang om patientbeskyttelse og behovet for tredjepartsgodkendelse smittede direkte af på den måde, man senere designede reglerne for medicinsk udstyr, især for produkter med høj risiko.
Hvad er 'Den Nye Tilgang' (the 'New Approach')?
Det er en EU-reguleringsfilosofi fra 1985, der skulle fremme det indre marked. I stedet for at lovgivningen detaljeret beskriver alle tekniske specifikationer, fastsætter den kun de overordnede, 'essentielle krav' til sikkerhed og ydeevne. Producenter kan derefter bruge harmoniserede tekniske standarder til at bevise, at de opfylder disse krav. Overensstemmelsen verificeres af et uafhængigt 'Bemyndiget organ', hvilket fører til den endelige CE-mærkning, der giver adgang til hele EU-markedet.
Var industrien imod regulering?
Overraskende nok, nej. Især for producenter af højrisikoudstyr som pacemakere var det kaotiske og uforudsigelige system med forskellige nationale regler en forretningsmæssig ulempe. Et samlet, forudsigeligt og harmoniseret europæisk regelsæt blev set som en fordel. Derfor var industriorganisationer som IAPM ikke blot passive modtagere af regulering, men aktive deltagere, der leverede ekspertise og forslag til Europa-Kommissionen for at forme de nye love.
Hvis du vil læse andre artikler, der ligner Vejen til sikkerhed: EU's udstyrsregulering, kan du besøge kategorien Sundhed.