05/05/2016
I den stærkt regulerede verden af farmaceutisk fremstilling og kvalitetskontrol er det afgørende at forstå og overholde reglerne for God Fremstillingspraksis (GMP). For laboratorier, der opererer globalt eller leverer produkter til forskellige markeder, er det en absolut nødvendighed at kende forskellene mellem kravene fra den amerikanske Food and Drug Administration (FDA) og det Europæiske Lægemiddelagentur (EMA). Selvom begge agenturer håndhæver strenge standarder for at sikre produktsikkerhed og -kvalitet, adskiller deres tilgange, forventninger og dokumentationskrav sig markant. Denne artikel giver en dybdegående vejledning til laboratorieledere, QA/QC-professionelle og regulatoriske specialister, der sammenligner FDA's og EMA's rammer og hjælper laboratorier med at forberede sig til inspektioner, undgå faldgruber og opretholde global markedsberedskab.

- Hvad er God Fremstillingspraksis (GMP)?
- FDA vs. EMA: En Forskel i Filosofi og Stil
- Inspektioner: Hvad Kigges Der Efter?
- Dokumentation og Opbevaring af Optegnelser
- Krav til Uddannelse for GMP-overholdelse
- Kvalificering af Leverandører: FDA vs. EMA Standarder
- Ofte Stillede Spørgsmål (FAQ)
- Konklusion: Harmonisering af FDA og EMA GMP for Globale Laboratorier
Hvad er God Fremstillingspraksis (GMP)?
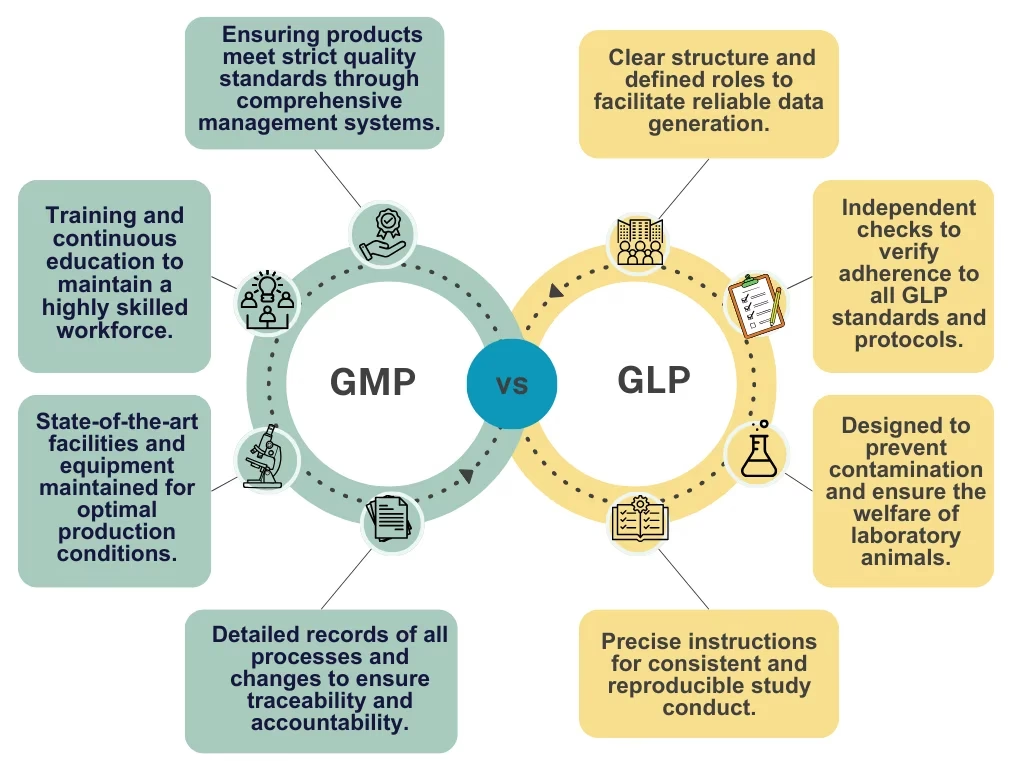
God Fremstillingspraksis (GMP) er et system, der sikrer, at produkter som lægemidler, kosmetik og fødevarer produceres og kontrolleres konsekvent i henhold til kvalitetsstandarder. Det er designet til at minimere de risici, der er involveret i enhver farmaceutisk produktion, som ikke kan elimineres gennem testning af det endelige produkt. Hovedformålet med GMP er at forhindre skade på slutbrugeren. GMP dækker alle aspekter af produktionen; fra startmaterialer, lokaler og udstyr til uddannelse og personlig hygiejne hos personalet. Detaljerede, skriftlige procedurer er afgørende for hver proces, der kan påvirke kvaliteten af det færdige produkt. Mange lande har lovgivet, at medicinal- og medicinsk udstyrsvirksomheder skal følge GMP-procedurer, og har skabt deres egne GMP-retningslinjer, der svarer til deres lovgivning. Du vil ofte se udtrykket 'cGMP', hvor 'c' står for 'current' (aktuel), hvilket understreger, at forventningerne til systemer og teknologi er dynamiske og løbende skal opdateres for at overholde den nyeste standard.
FDA vs. EMA: En Forskel i Filosofi og Stil
Selvom både FDA og EMA har det fælles mål at sikre produkters sikkerhed, effektivitet og kvalitet, er deres grundlæggende filosofier forskellige. At forstå denne forskel er nøglen til at navigere i begge regelsæt med succes.
FDA's Tilgang – Præskriptiv og Regelbaseret
FDA's GMP-regler, som er kodificeret i 21 CFR del 210 og 211, er yderst detaljerede og præskriptive. Det betyder, at reglerne specificerer præcist, hvad der skal gøres, og hvordan det skal gøres. Laboratorier og produktionsfaciliteter skal nøje følge disse foruddefinerede protokoller. Afvigelser kan hurtigt føre til en observationsrapport (Form 483) eller en advarselsskrivelse, som kan have alvorlige konsekvenser for en virksomheds drift og omdømme. Denne tilgang efterlader mindre plads til fortolkning, men kræver en omhyggelig og stringent overholdelse af de skrevne regler.
EMA's Tilgang – Direktiv- og Principbaseret
I modsætning hertil er EMA's regler, især EudraLex bind 4, mere principbaserede. I stedet for at diktere hver eneste detalje, lægger EMA vægt på implementeringen af et robust kvalitetsstyringssystem (QMS) og en integreret tilgang til kvalitetsrisikostyring. EMA forventer, at virksomheder fortolker de overordnede principper og implementerer systemer, der er i overensstemmelse med disse, understøttet af solid dokumentation og en klar begrundelse baseret på risikovurdering. Dette giver virksomheder mere fleksibilitet, men kræver samtidig en dybere forståelse af de underliggende kvalitetsprincipper.

Sammenlignende Oversigt
Nedenstående tabel opsummerer de centrale forskelle i de to agenturers regulatoriske stil:
| Aspekt | FDA (USA) | EMA (EU) |
|---|---|---|
| Regulatorisk Stil | Præskriptiv (regelbaseret) i henhold til 21 CFR Parts 210/211. | Direktiv-baseret (principbaseret) i henhold til EudraLex Vol. 4. |
| Kvalitetsrisikostyring | Valgfrit indtil for nylig, nu mere integreret. | Påkrævet og centralt element under ICH Q9-vejledningen. |
| Fokus på QMS | Til stede, men ikke så formaliseret som hos EMA. | Stærkt fokus på et omfattende farmaceutisk QMS. |
| Opdateringer | Langsommere til at adoptere nye internationale vejledninger (ICH). | Hurtig inkorporering af ICH-opdateringer i regelsættet. |
Inspektioner: Hvad Kigges Der Efter?
Regulatoriske inspektioner er kritiske begivenheder med høje indsatser. At vide, hvad hvert agentur fokuserer på, kan hjælpe laboratorier med at forberede sig og undgå manglende overholdelse.
FDA-inspektørers Fokusområder:
- Dataintegritet og ALCOA+ principperne (Attributable, Legible, Contemporaneous, Original, Accurate). Inspektører vil granske rådata, audit trails og elektroniske optegnelser for at sikre, at data er pålidelige og ikke er blevet manipuleret.
- Specifikke fremstillingsprocesser og afvigelser. De vil sammenligne de faktiske processer med de godkendte skriftlige procedurer (SOP'er) og undersøge enhver afvigelse i detaljer.
- Dokumentationens sporbarhed og batchoptegnelser. Det skal være muligt at spore hvert trin i en batchs livscyklus fra råmateriale til færdigt produkt.
EMA-inspektørers Fokusområder:
- Systemdækkende kvalitets-risikostyring. Inspektører vil vurdere, hvordan virksomheden identificerer, analyserer og håndterer risici på tværs af hele organisationen.
- Validerings- og kvalificeringslivscyklus. De ser på den samlede strategi for validering af processer, udstyr og metoder, ikke kun den endelige rapport.
- Integration af kvalitetsstyringssystemer (QMS). De undersøger, hvordan systemer som afvigelseshåndtering, CAPA (Corrective and Preventive Actions) og change control arbejder sammen som en sammenhængende enhed.
Dokumentation og Opbevaring af Optegnelser
Både FDA og EMA understreger vigtigheden af grundig dokumentation for at sikre gennemsigtighed, sporbarhed og overholdelse. Deres forventninger til format, dybde og opbevaring varierer dog betydeligt.
FDA's Forventninger:
- Samidig registrering: Data skal registreres på det tidspunkt, hvor aktiviteten udføres. For eksempel skal pH-målinger logges direkte i en laboratoriejournal eller et valideret elektronisk system, ikke på et løst stykke papir til senere overførsel.
- Rådata og underskrifter: Originale laboratoriedata (f.eks. kromatogrammer) skal opbevares med analytikerens underskrift og dato for review for at sikre sporbarhed.
- Opbevaringsperiode: FDA kræver typisk, at optegnelser opbevares i mindst 1 år efter produktets udløbsdato.
EMA's Forventninger:
- QMS-integration: Dokumentation forventes at være tæt forbundet med et kvalitetsstyringssystem. SOP'er, afvigelsesrapporter og CAPA-dokumentation skal afspejle en risikobaseret, systemisk tilgang.
- Kontrolleret dokumentation: EMA kræver streng versionskontrol, hierarkier for dokumentgodkendelse og audit trails. Et eksempel er at vedligeholde en ændringslog, der sporer, hvem der reviderede en SOP, hvornår og hvorfor.
- Opbevaringsperiode: EMA kræver normalt, at optegnelser opbevares i mindst 5 år efter frigivelse af batchen. For biologiske og sterile produkter kan denne tidslinje være endnu længere.
Krav til Uddannelse for GMP-overholdelse
Personalets kompetence er et hyppigt fokuspunkt under inspektioner. Mens begge myndigheder kræver træning, går EMA et skridt videre ved at kræve en dybere integration med QMS.
- FDA Retningslinjer: Periodisk GMP-træning er obligatorisk. Rollebaseret træning skal spores via træningsfiler for hver medarbejder.
- EMA Retningslinjer: Træning skal være en integreret del af QMS. Der er et krav om løbende evaluering af træningens effektivitet for at sikre, at personalet ikke kun har deltaget, men også har forstået og kan anvende viden i praksis.
Kvalificering af Leverandører: FDA vs. EMA Standarder
Globaliseringen af den farmaceutiske forsyningskæde har skærpet tilsynet med råvarekilder. Her er, hvordan FDA og EMA håndterer leverandørkvalificering:
| Kriterium | FDA Krav | EMA Krav |
|---|---|---|
| Leverandøraudits | Anbefales, men er ikke altid obligatorisk. | Påkrævet for alle kritiske leverandører. |
| Materialetestning | Fuld testning af indgående materialer, medmindre reduceret testning er valideret. | Risikobaseret tilgang styret af QMS. |
| Kvalificeringsoptegnelser | Fremhævet i 21 CFR 211.84. | Integreret i Annex 8 og Annex 16. |
Ofte Stillede Spørgsmål (FAQ)
Hvad er de største forskelle mellem FDA og EMA GMP-regler?
Den primære forskel ligger i tilgangen. FDA anvender en regelbaseret, præskriptiv tilgang (21 CFR 210/211), der specificerer 'hvordan'. EMA anvender en principbaseret tilgang (EudraLex Vol. 4), der fokuserer på 'hvad' der skal opnås gennem et robust kvalitetsstyringssystem (QMS) og kvalitetsrisikostyring (QRM).
Hvordan skal et laboratorium forberede sig til både FDA- og EMA-inspektioner?
Den bedste strategi er at implementere et samlet QMS, der opfylder de strengeste krav fra begge agenturer. Udfør regelmæssige gapanalyser, gennemfør interne 'mock' inspektioner baseret på begge regelsæt, og sørg for omfattende dokumentation og træning, der dækker begge standarder.
Er tidsfristerne for opbevaring af optegnelser forskellige?
Ja. FDA kræver typisk opbevaring i 1 år efter produktets udløbsdato. EMA kræver opbevaring i mindst 5 år efter batch-frigivelse, og potentielt længere for visse produkttyper.
Hvad er den bedste strategi for en virksomhed, der opererer på begge markeder?
Den sikreste og mest effektive strategi er at identificere de strengeste krav for hvert specifikt område (f.eks. dokumentationsopbevaring, leverandøraudits) og implementere disse på tværs af hele organisationen. Dette skaber et enkelt, robust kvalitetssystem, der er i overensstemmelse med begge regelsæt og minimerer risikoen for mangler under inspektioner.
Konklusion: Harmonisering af FDA og EMA GMP for Globale Laboratorier
For laboratorier, der leverer til både det amerikanske og europæiske marked, er det afgørende at forstå de nuancerede forskelle mellem FDA's og EMA's GMP-regler. Fra dokumentationspraksis til træningsstrategier og leverandørkvalificering sikrer en tilpasning til begge regelsæt ikke kun overholdelse, men forbedrer også produktkvaliteten og markedstilliden. GMP handler ikke kun om at afkrydse bokse – det er et fundamentalt lag af videnskabelig stringens og operationel excellence. Ved at forblive informeret kan laboratorieprofessionelle og ledere strømline inspektioner, minimere risici og styrke deres globale konkurrenceevne.
Hvis du vil læse andre artikler, der ligner FDA vs. EMA: Guide til GMP-regler for Lægemidler, kan du besøge kategorien Sundhed.