20/08/2002
Reglerne og retningslinjerne, der styrer driften af kliniske laboratorier, udvides og opdateres konstant. Den amerikanske Fødevare- og Lægemiddeladministration (FDA) er ved at udfase sin håndhævelsesdiskretion over for laboratorieudviklede tests (LDT'er), Clinical Laboratory Improvement Amendments (CLIA) har etableret nye kvalitetsstandarder for laboratoriepersonale og færdighedstest, og selv Den Internationale Standardiseringsorganisation (ISO) opdaterer sine krav mindst en gang hvert femte år. Med så mange ændringer på én gang, hvordan kan laboratorier sikre, at de holder sig opdaterede og overholder reglerne? Denne artikel vil dykke ned i kompleksiteten af nutidens regulatoriske miljø for kliniske laboratorier og give klarhed over de vigtigste forskelle, overlapninger og fremtidige ændringer, som alle laboratorieprofessionelle bør kende til.

Forståelse af de Vigtigste Reguleringsorganer: FDA, CLIA og ISO
For at navigere i det regulatoriske landskab er det afgørende først at forstå de centrale aktører og deres respektive roller. Selvom de alle sigter mod at sikre kvalitet og sikkerhed i laboratorietests, er deres fokus, autoritet og anvendelsesområde markant forskellige.
FDA (Food and Drug Administration)
FDA er en føderal regulerende myndighed i USA. Deres primære rolle i denne sammenhæng er at regulere medicinsk udstyr, herunder in vitro-diagnostiske (IVD) tests, som anvendes i kliniske laboratorier. FDA er ansvarlig for at godkende disse tests, før de kan markedsføres, og de kategoriserer også testenes kompleksitet (f.eks. undtagne, moderat komplekse, højt komplekse). Indtil for nylig havde kliniske laboratorier, der udviklede deres egne tests (LDT'er), ikke været underlagt mange FDA-regler. Dette er dog ved at ændre sig drastisk med introduktionen af en ny endelig regel, der betragter disse laboratorier som producenter, hvilket underlægger dem FDA's krav til sikkerhed og effektivitet.
CLIA (Clinical Laboratory Improvement Amendments)
CLIA er et sæt obligatoriske føderale regler, der gælder specifikt for kliniske laboratorier i USA. Et laboratorium skal være CLIA-certificeret af Centers for Medicare & Medicaid Services (CMS) for at kunne modtage humane prøver til testning. CLIA-kravene fokuserer på at opretholde høje kvalitetsstandarder i alle aspekter af laboratoriedriften. Dette omfatter alt fra prøveindsamling, kvalitetskontrolprocedurer og rapportering af resultater til personkvalifikationer, uddannelse og kompetencevurdering. I USA er CLIA den centrale myndighed, da de er de primære administratorer af reglerne, herunder udførelse af laboratorieinspektioner og håndhævelse af overholdelse.
ISO (International Organization for Standardization)
I modsætning til FDA og CLIA er ISO en international, frivillig organisation. De udarbejder retningslinjer, der ikke er juridisk bindende, medmindre de er blevet vedtaget i et lands lovgivningsmæssige rammer. For kliniske laboratorier er ISO 15189 den relevante standard, der specificerer krav til kvalitet og kompetence. At opnå ISO-certificering er valgfrit for amerikanske laboratorier, ligesom akkreditering fra College of American Pathologists (CAP). Mange laboratorier vælger dog at forfølge disse certificeringer for at demonstrere deres engagement i kvalitet og kontinuerlig forbedring, hvilket kan være påkrævet for specifikke kontrakter eller internationalt arbejde.
Sammenligning af Reguleringsorganernes Ansvarsområder
For at skabe et klart overblik over, hvordan disse tre organer adskiller sig, kan en sammenlignende tabel være nyttig.
| Egenskab | FDA | CLIA | ISO |
|---|---|---|---|
| Type | Føderal regulerende myndighed (USA) | Obligatoriske føderale regler (USA) | International, frivillig standardiseringsorganisation |
| Primært Fokus | Regulering af medicinsk udstyr, inkl. IVD-tests. Sikkerhed og effektivitet af produkter. | Kvalitetsstandarder for laboratoriedrift, personale og processer. | Krav til kvalitet og kompetence i medicinske laboratorier (ISO 15189). |
| Anvendelse | Obligatorisk for producenter af medicinsk udstyr. Nu også for laboratorier, der udvikler LDT'er. | Obligatorisk for alle laboratorier i USA, der udfører tests på humane prøver. | Frivillig, men kan være påkrævet for kontrakter eller international anerkendelse. |
| Håndhævelse | Håndhæves af FDA. | Håndhæves primært af CMS, som udsteder certifikater og udfører inspektioner. | Certificering udføres af akkrediterede tredjepartsorganer. |
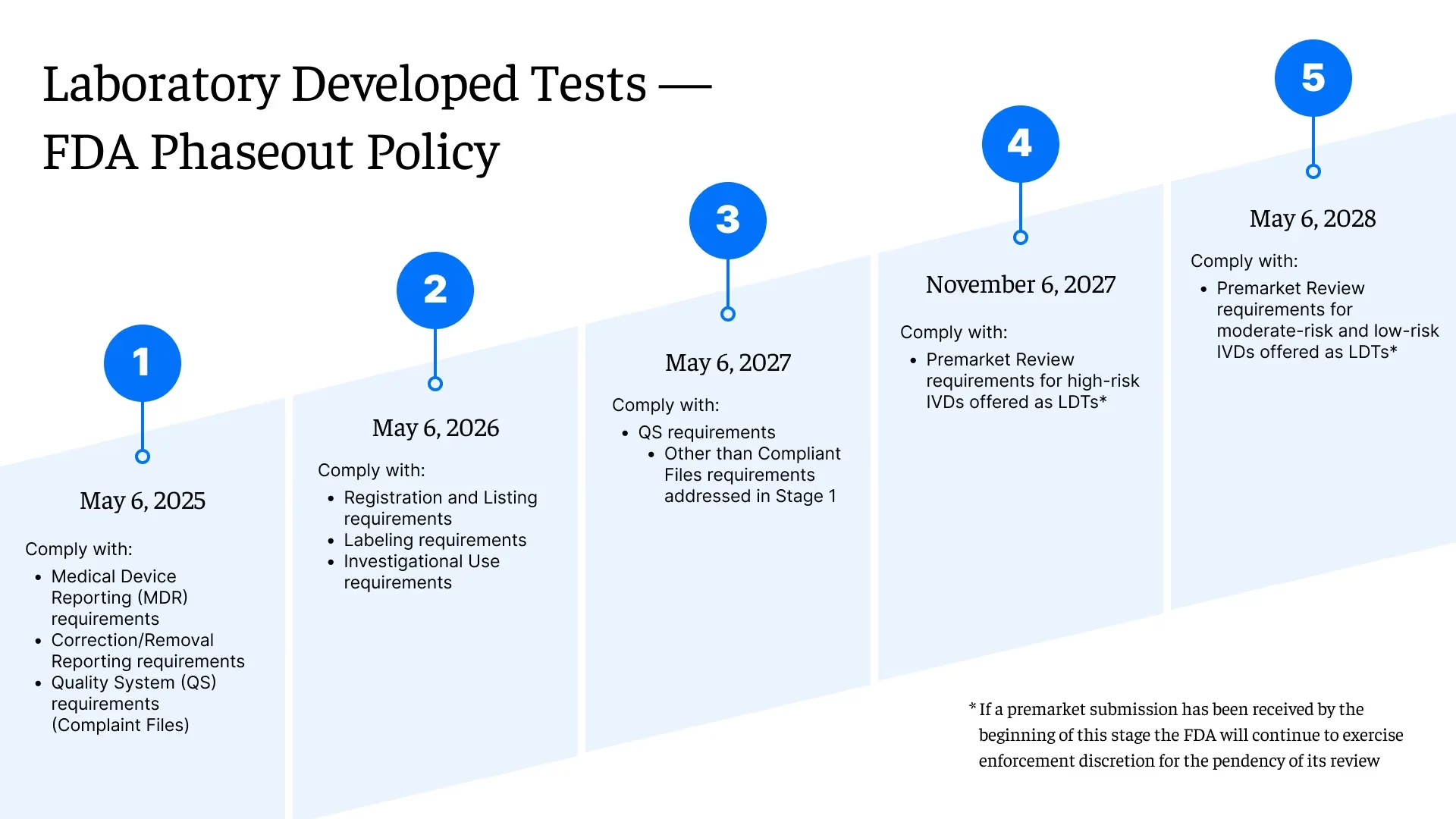
Den Nye FDA-Regel for LDT'er: En Game-Changer
Den mest betydningsfulde nylige ændring er FDA's endelige regel om regulering af laboratorieudviklede tests (LDT'er). Historisk set har laboratorier, der udviklede og udførte deres egne tests internt, opereret under en politik med "håndhævelsesdiskretion" fra FDA. Den nye regel ændrer dette fundamentalt ved at klassificere disse laboratorier som producenter af medicinsk udstyr.
Hvad betyder det i praksis? Laboratorier, der tilbyder LDT'er og ikke falder ind under nogen af undtagelserne i den endelige regel, skal nu overholde FDA's regler. Dette indebærer, at de skal bevise sikkerheden og effektiviteten af deres tests på en måde, der ligner kommercielle IVD-producenter. Denne overgang vil ske i faser over de næste flere år. Det er afgørende at understrege, at disse nye FDA-regler kommer i tillæg til, ikke i stedet for, de eksisterende CLIA-krav. Laboratorier skal overholde begge regelsæt, hvilket skaber et mere komplekst og potentielt dobbeltarbejdende regulatorisk miljø.
Udfordringer og Overlapninger i det Nye Landskab
Med indførelsen af FDA's LDT-regel opstår der nye udfordringer, især inden for områder, hvor reglerne overlapper eller er dobbelte.
- Kvalitetssystemer: Laboratorier overholder allerede kvalitetsregler under CLIA. Nu skal de også opfylde yderligere krav fra FDA, som er tæt afstemt med ISO 13485. Selvom koncepterne er ens, er terminologien ofte forskellig, hvilket skaber en stejl læringskurve.
- Valideringsstudier: Hvor CLIA giver anbefalinger til validering, kan FDA være mere specifik og kræve overholdelse af bestemte retningslinjer fra organisationer som Clinical & Laboratory Standards Institute (CLSI).
- Rapportering af uønskede hændelser: Et eksempel på dobbeltarbejde er rapportering af alvorlige hændelser. Et laboratorium, hvis LDT kan have forårsaget en alvorlig skade, kan nu være forpligtet til at rapportere hændelsen til FDA både som "bruger" (under eksisterende regler) og som "producent" (under den nye regel). Detaljerne omkring implementeringen er stadig under udvikling.
Den største udfordring for mange laboratorier vil være kravet om designkontrol. Dette er en central del af FDA's regler for producenter, men det er et koncept, som de færreste laboratorier har erfaring med fra CLIA, der primært fokuserer på laboratorieprocesser og personale.
Ofte Stillede Spørgsmål (FAQ)
Hvad er den primære forskel mellem FDA, CLIA og ISO?
Kort sagt regulerer FDA produkterne (f.eks. IVD-tests), CLIA regulerer laboratoriets drift og kvalitet, og ISO er en frivillig international standard for kvalitet og kompetence. FDA og CLIA er obligatoriske i USA, mens ISO er valgfrit.
Er ISO-certificering obligatorisk for kliniske laboratorier?
Nej, ISO-certificering (f.eks. ISO 15189) er ikke et lovkrav for kliniske laboratorier i USA. Det er en frivillig akkreditering, som laboratorier kan vælge at opnå for at demonstrere et højt niveau af kvalitet og forbedre deres processer, hvilket kan være en fordel i visse forretningssammenhænge.
Hvad er den største ændring i FDA's regulering af laboratorier?
Den største ændring er den nye endelige regel for laboratorieudviklede tests (LDT'er). Denne regel fjerner den tidligere håndhævelsesdiskretion og behandler laboratorier, der udvikler deres egne tests, som producenter af medicinsk udstyr, hvilket kræver, at de overholder FDA's regler for sikkerhed og effektivitet.
Erstatter de nye FDA-regler for LDT'er de eksisterende CLIA-krav?
Nej, absolut ikke. Dette er en almindelig misforståelse. FDA's regler er et supplement til, ikke en erstatning for, CLIA-kravene. Laboratorier, der er omfattet af den nye regel, skal overholde begge regelsæt. Dette øger den samlede regulatoriske byrde.
Hvor kæmper laboratorier mest med overholdelse?
Mange laboratorier står over for udfordringer på grund af mangel på personale og ekspertise til fuldt ud at forstå og implementere de nye FDA-krav, især oven i de eksisterende CLIA-forpligtelser. Det mest udfordrende nye område forventes at være designkontrol, som er et centralt element i FDA's krav, men som er ukendt for de fleste laboratorier, der kun har arbejdet under CLIA.
Konklusion: Forberedelse på Fremtiden
Kliniske laboratorier står over for en perfekt storm af forestående regulatoriske ændringer. At navigere i dette komplekse samspil mellem FDA, CLIA og endda ISO-standarder kræver proaktivitet og en dyb forståelse af kravene. Den nye FDA-regel for LDT'er repræsenterer et paradigmeskifte, der vil kræve betydelige ressourcer, tid og ekspertise at implementere. For at fortsætte med at imødekomme patienternes behov er det afgørende for laboratorier at holde sig foran kurven, søge vejledning og investere i den nødvendige viden for at sikre fortsat overholdelse og levere tests af højeste kvalitet.
Hvis du vil læse andre artikler, der ligner FDA, CLIA & ISO: Guide til Laboratorie-Regler, kan du besøge kategorien Sundhed.