
06/02/2024
Vejen fra en forskers spæde idé til et nyt lægemiddel, der kan redde liv, er en utrolig kompleks, tidskrævende og ofte årelang proces. Det kræver omfattende forskning, strenge tests og et tæt samarbejde mellem videnskabsfolk, farmaceutiske virksomheder og regulatoriske myndigheder. Hvert eneste lægemiddel på apotekets hylder har gennemgået denne maraton, hvor kun en brøkdel af de oprindelige kandidater når i mål. Denne artikel vil guide dig gennem de afgørende faser i lægemiddeludvikling, fra det første eureka-øjeblik i laboratoriet til den færdige pille i patientens hånd.

- Den Første Gnist: Idéen og Opdagelsen
- Præklinisk Forskning: Sikkerhed Først
- De Kliniske Faser: Den Ultimative Test på Mennesker
- En Sammenligning af de Kliniske Faser
- Godkendelsesprocessen: Det Sidste Nåleøje
- Fra Fabrik til Apotek: Produktion og Overvågning
- Omkostninger, Tid og Patenter: Den Økonomiske Virkelighed
- Ofte Stillede Spørgsmål om Udvikling af Lægemidler
Den Første Gnist: Idéen og Opdagelsen
Alt begynder med en idé. Inspirationen til ny medicin opstår ofte fra en dyb forståelse af sygdomme på et molekylært niveau. Forskere undersøger, hvordan specifikke kemiske forbindelser kan interagere med kroppens biologiske systemer for at bekæmpe en sygdom. Denne indledende fase involverer grundige litteraturstudier, laboratorieforskning og innovativ tænkning for at identificere potentielle terapeutiske "mål" – typisk et protein eller et gen, der spiller en central rolle i sygdomsprocessen.
Moderne teknologi har revolutioneret denne opdagelsesfase. Med fremskridt inden for genomik og bioteknologi kan forskere nu identificere helt nye mål for lægemidler. Ved hjælp af metoder som high-throughput screening kan tusindvis af kemiske forbindelser testes mod et specifikt biologisk mål på meget kort tid. Ud af måske 10.000 testede forbindelser udvælges kun en håndfuld, der viser lovende potentiale for videre undersøgelse.
Præklinisk Forskning: Sikkerhed Først
Når en lovende kemisk forbindelse er identificeret, begynder den prækliniske forskning. Denne fase omfatter en række laboratorie- og dyreforsøg, der har til formål at vurdere lægemiddelkandidatens sikkerhed, effektivitet og farmakokinetik – altså hvordan stoffet opfører sig i kroppen. Forskerne undersøger, hvordan stoffet absorberes, fordeles, metaboliseres og udskilles. Denne viden er afgørende for at kunne bestemme en passende dosis og administrationsmåde for de senere forsøg på mennesker.
Det er et lovkrav, at der udføres en grundig vurdering af stoffets potentielle giftighed for kroppens vigtigste organer, herunder hjerte, lunger, hjerne, nyrer og lever. Selvom mange indledende tests kan udføres in vitro (i reagensglas med isolerede celler), er dyreforsøg ofte nødvendige for at forstå det komplekse samspil mellem stofskifte og toksicitet i en levende organisme. Samtidig arbejdes der med formuleringen af lægemidlet for at skabe et stabilt og effektivt produkt, hvad enten det skal være en tablet, kapsel eller en injektionsvæske.
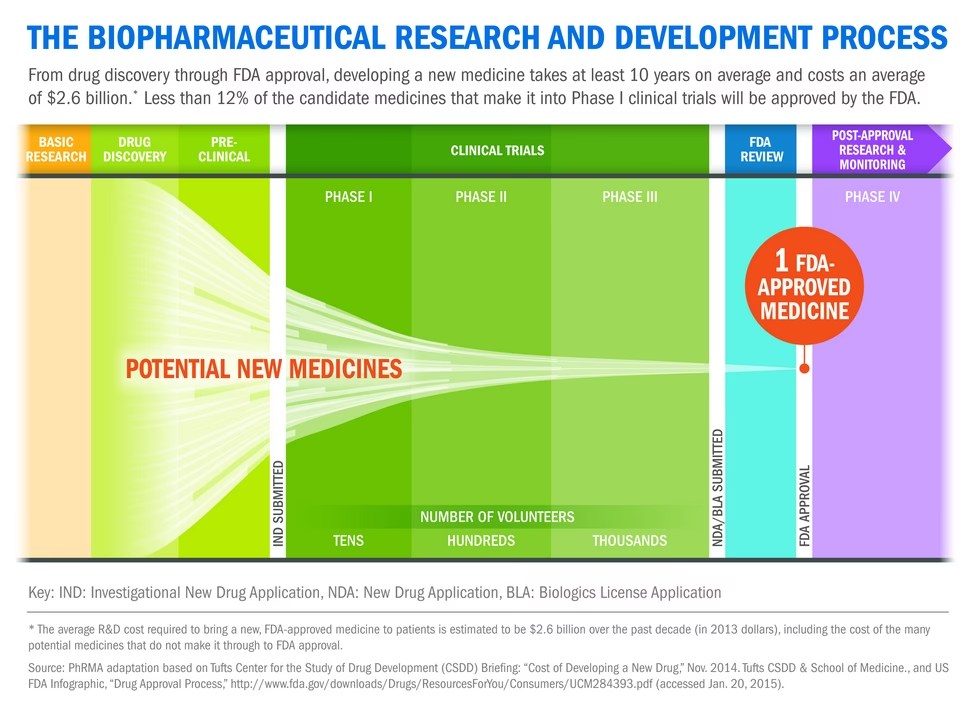
De Kliniske Faser: Den Ultimative Test på Mennesker
Hvis de prækliniske studier viser lovende resultater og en acceptabel sikkerhedsprofil, kan lægemiddelkandidaten gå videre til kliniske forsøg på mennesker. Denne proces er strengt reguleret og kræver godkendelse fra nationale lægemiddelmyndigheder, som f.eks. Lægemiddelstyrelsen i Danmark. Kliniske forsøg er typisk opdelt i tre hovedfaser, før en eventuel godkendelse.
Fase I: Er det sikkert?
Den første fase involverer en lille gruppe (typisk 20-100) raske, frivillige forsøgspersoner. Hovedformålet er at vurdere lægemidlets sikkerhed i mennesker, fastlægge et sikkert dosisinterval og identificere eventuelle bivirkninger. Deltagerne overvåges nøje, og der indsamles detaljerede data om, hvordan kroppen håndterer stoffet. Omkring halvdelen af de kandidater, der når hertil, betragtes som sikre nok til at fortsætte til næste fase.
Fase II: Virker det?
I fase II gives lægemidlet til en større gruppe patienter (100-500), som lider af den sygdom, medicinen er rettet imod. Formålet her er at få en indledende vurdering af lægemidlets effektivitet og yderligere undersøge dets sikkerhed. Forskerne sigter mod at finde den mest effektive dosis og administrationsmetode. Mange lægemiddelkandidater fejler i denne fase, enten fordi de viser sig at være ineffektive, eller fordi de har uacceptable bivirkninger.
Fase III: Er det bedre end det eksisterende?
Dette er den sidste og mest omfattende fase før en eventuel markedsføringstilladelse. Fase III-forsøg involverer tusindvis af patienter (ofte 1.000-5.000) på tværs af flere lande. Her sammenlignes det nye lægemiddel med eksisterende standardbehandlinger eller placebo (en inaktiv behandling). Målet er at indsamle robuste data, der endeligt kan bekræfte lægemidlets effektivitet og sikkerhed i en stor og forskelligartet patientpopulation. Disse data danner grundlaget for ansøgningen om godkendelse hos myndighederne.
En Sammenligning af de Kliniske Faser
| Fase | Primært Formål | Antal Deltagere | Deltagertype |
|---|---|---|---|
| Fase I | Vurdere sikkerhed og dosering | 20-100 | Raske frivillige |
| Fase II | Vurdere effektivitet og bivirkninger | 100-500 | Patienter med sygdommen |
| Fase III | Bekræfte effektivitet, overvåge sikkerhed, sammenligne med standardbehandling | 1.000-5.000+ | Patienter med sygdommen |
Godkendelsesprocessen: Det Sidste Nåleøje
Efter vellykkede kliniske forsøg indsender medicinalfirmaet en ansøgning om markedsføringstilladelse til de relevante regulatoriske myndigheder. I Europa sker dette ofte centralt via Det Europæiske Lægemiddelagentur (EMA), hvilket giver adgang til hele EU-markedet. I USA er det Food and Drug Administration (FDA), der står for godkendelsen. Ansøgningen er et massivt dokument, der indeholder alle data fra prækliniske og kliniske studier, information om fremstillingsprocessen, kvalitetskontrol og forslag til produktinformation.
Myndighedernes eksperter gennemgår omhyggeligt alle data for at vurdere lægemidlets kvalitet, sikkerhed og effekt. De foretager en grundig afvejning af fordele over for risici. Kun hvis fordelene klart opvejer risiciene, vil lægemidlet blive godkendt og få tilladelse til at blive solgt. Denne strenge proces er designet til at beskytte folkesundheden og sikre, at kun sikre og effektive lægemidler når markedet.
Fra Fabrik til Apotek: Produktion og Overvågning
Når et lægemiddel er godkendt, starter produktionen i stor skala. Fabrikkerne skal overholde strenge regler kendt som Good Manufacturing Practices (GMP) for at sikre ensartet kvalitet og renhed i hver eneste batch. Kvalitetskontrol er en integreret del af hele produktionsprocessen.
Men rejsen slutter ikke her. Selv efter at medicinen er kommet på markedet, fortsætter overvågningen. Dette kaldes Fase IV eller post-market surveillance. Formålet er at indsamle data om langsigtede effekter og identificere sjældne bivirkninger, som måske ikke blev opdaget i de kliniske forsøg. Læger, patienter og medicinalfirmaer indberetter eventuelle uventede hændelser, hvilket sikrer en kontinuerlig overvågning af lægemidlets sikkerhedsprofil.
Omkostninger, Tid og Patenter: Den Økonomiske Virkelighed
Udviklingen af et nyt lægemiddel er en enorm investering. Hele processen fra idé til marked tager i gennemsnit 10-15 år og kan koste over 10 milliarder kroner. En af de primære årsager til de høje omkostninger er den ekstremt høje fejlrate. For hver 25.000 forbindelser, der starter i laboratoriet, når kun omkring 25 til test på mennesker, og kun fem ender med at blive godkendt. Kun én af disse fem vil typisk tjene nok til at dække udviklingsomkostningerne for sig selv og alle de fejlslagne projekter.
For at give virksomhederne et incitament til at påtage sig denne enorme økonomiske risiko, beskyttes nye lægemidler af et patent. Et patent giver virksomheden eneret til at markedsføre lægemidlet i en periode på typisk 20 år fra ansøgningsdatoen. Når patentet udløber, kan andre virksomheder producere og sælge billigere generiske versioner af medicinen.
Ofte Stillede Spørgsmål om Udvikling af Lægemidler
Hvorfor tager det så lang tid at udvikle ny medicin?
Processen er lang, fordi hvert trin er designet til at sikre patienternes sikkerhed og lægemidlets effektivitet. De prækliniske tests og de tre faser af kliniske forsøg er alle tidskrævende, men nødvendige for at indsamle tilstrækkelige data til en grundig vurdering.

Hvorfor er ny medicin ofte så dyr?
De høje priser afspejler de enorme omkostninger til forskning og udvikling, herunder udgifterne til de mange tusinde lægemiddelkandidater, der fejler undervejs. Patentbeskyttelsen giver virksomheden en tidsbegrænset mulighed for at tjene sin investering hjem og finansiere fremtidig forskning.
Hvem godkender medicin i Danmark?
I Danmark er det Lægemiddelstyrelsen, der godkender og overvåger lægemidler. For mange nye lægemidler sker godkendelsen dog centralt for hele EU via Det Europæiske Lægemiddelagentur (EMA), hvorefter godkendelsen også gælder i Danmark.
Hvad sker der, når et lægemiddels patent udløber?
Når patentet udløber, kan andre medicinalfirmaer producere og sælge såkaldte generiske lægemidler. Disse indeholder det samme aktive stof og har samme virkning som det originale produkt, men er typisk meget billigere, da producenterne ikke har haft de samme udviklingsomkostninger.
Hvis du vil læse andre artikler, der ligner Fra Idé til Pille: Rejsen for et Nyt Lægemiddel, kan du besøge kategorien Sundhed.