01/08/2009
Cystisk fibrose (CF) er en kompleks og kronisk arvelig sygdom, der primært påvirker lungerne og fordøjelsessystemet ved at forårsage produktion af tykt, sejt slim. Heldigvis har behandlingsmulighederne udviklet sig markant i de seneste årtier, hvilket har forbedret både livskvaliteten og levetiden for patienter. Moderne behandling fokuserer ikke kun på at håndtere symptomer, men også på at korrigere den underliggende årsag til sygdommen på et molekylært niveau. Denne artikel giver et dybdegående indblik i de forskellige medicinske tilgange til behandling af cystisk fibrose.

Grundlæggende Behandlingsstrategier
Behandlingen af cystisk fibrose er mangesidet og kræver en personlig tilgang, der er skræddersyet til den enkelte patient. Målene for behandlingen er generelt at vedligeholde og forbedre lungefunktionen, bekæmpe infektioner, fjerne det seje slim fra luftvejene og forbedre den generelle ernæringstilstand. Dette opnås gennem en kombination af medicin, fysioterapi (lungefysioterapi) og ernæringsstøtte.
Medicin til Symptomlindring og Infektionsbekæmpelse
En stor del af den daglige behandling for CF-patienter involverer medicin, der sigter mod at lindre symptomer og forebygge de alvorlige komplikationer, der kan opstå, især i lungerne.
Antibiotika: Den konstante kamp mod infektioner
Lungerne hos personer med CF er særligt sårbare over for kroniske bakterielle infektioner på grund af det tykke slim, hvor bakterier trives. Antibiotika er derfor en hjørnesten i behandlingen. De bruges både til at forebygge og behandle lungeinfektioner, hvilket er afgørende for at bevare lungefunktion. Afhængigt af infektionens art og sværhedsgrad kan lægen ordinere:
- Oral antibiotika: Tabletter eller mikstur, der tages derhjemme for at behandle milde til moderate infektioner.
- Inhaleret antibiotika: En forstøver bruges til at levere medicinen direkte til lungerne. Dette er en effektiv måde at opnå høje koncentrationer af medicin i luftvejene med færre systemiske bivirkninger.
- Intravenøs (IV) antibiotika: Ved alvorlige lungeinfektioner kan det være nødvendigt med hospitalsindlæggelse for at modtage antibiotika direkte i blodbanen, hvilket sikrer en hurtig og kraftfuld effekt.
Antiinflammatoriske lægemidler
Kronisk inflammation er en central del af lungesygdommen ved CF og bidrager til nedbrydningen af lungevæv over tid. Antiinflammatoriske lægemidler bruges til at dæmpe denne betændelsesreaktion.
- Ibuprofen: I høje doser har ibuprofen vist sig at bremse faldet i lungefunktion, især hos børn. Behandlingen kræver dog tæt overvågning på grund af potentielle bivirkninger som mave- og nyreproblemer.
- Kortikosteroider: Disse kraftige betændelseshæmmende lægemidler kan bruges i perioder, men langvarig brug er begrænset på grund af risiko for bivirkninger som knogleskørhed, forhøjet blodsukker og forhøjet blodtryk.
Bronkodilatatorer og Slimløsnende Midler
For at gøre det lettere at trække vejret og fjerne slim fra lungerne, anvendes ofte inhalationsmedicin.
- Bronkodilatatorer: Disse lægemidler virker ved at afslappe musklerne omkring luftvejene, hvilket åbner dem op og gør vejrtrækningen lettere. De tages typisk via en inhalator før lungefysioterapi for at maksimere effekten.
- Slimløsnende midler (mukolytika): Disse medikamenter, som også inhaleres, gør det seje slim mere tyndtflydende og lettere at hoste op. Dette er afgørende for den daglige proces med at rense luftvejene.
Revolutionen inden for CF-behandling: CFTR-modulatorer
Den mest markante udvikling i behandlingen af cystisk fibrose er uden tvivl fremkomsten af CFTR-modulatorer. I modsætning til traditionel medicin, der kun behandler symptomerne, sigter disse lægemidler mod at korrigere den grundlæggende defekt forårsaget af mutationer i CFTR-genet. De virker ved at hjælpe det fejlbehæftede CFTR-protein med at fungere mere normalt.
Det er vigtigt at understrege, at CFTR-modulatorer ikke virker for alle med cystisk fibrose. Deres effektivitet afhænger af den specifikke genetisk mutation, patienten har. Derfor er en genetisk test nødvendig for at afgøre, om en patient er kandidat til denne type behandling. Disse lægemidler tages som tabletter og kan have en dramatisk positiv effekt på lungefunktion, vægtøgning og generel livskvalitet.
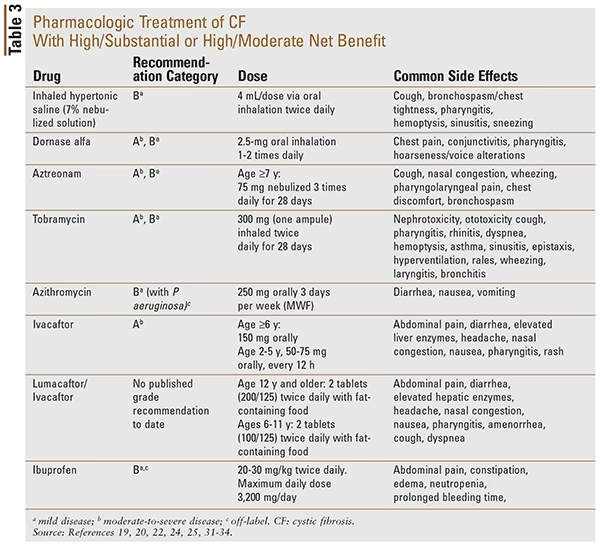
Der findes flere typer CFTR-modulatorer, ofte i kombination:
| Lægemiddel (Kombination) | Målgruppe (Alder og Mutationer) | Primær Virkning |
|---|---|---|
| Elexacaftor/tezacaftor/ivacaftor | Godkendt til voksne og børn fra 2 år og opefter med specifikke CFTR-mutationer. Dækker op til 90% af patientpopulationen. | En potent triple-kombination, der markant forbedrer funktionen af det defekte CFTR-protein. |
| Ivacaftor | Godkendt til voksne og børn helt ned til 4 måneder med visse "gating"-mutationer. | Forbedrer åbningen af CFTR-proteinkanalen på celleoverfladen. |
| Lumacaftor/ivacaftor | Godkendt til personer fra 1 år og opefter med to kopier af F508del-mutationen. | Hjælper med at transportere det defekte protein til celleoverfladen og forbedrer dets funktion. |
| Tezacaftor/ivacaftor | Godkendt til personer fra 4 måneder og opefter med specifikke mutationer, herunder F508del. | Ligner lumacaftor/ivacaftor, men har ofte færre bivirkninger og interaktioner med anden medicin. |
Selvom CFTR-modulatorer er revolutionerende, kan de have bivirkninger som midlertidig forværring af vejrtrækningssymptomer, hovedpine eller mavesmerter. I sjældne tilfælde kan der opleves angst eller depression. Det er også vigtigt at være opmærksom på, at visse etniske grupper har en højere forekomst af mutationer, der endnu ikke kan behandles med de nuværende modulatorer. For disse patienter er deltagelse i kliniske forsøg med nye lægemidler ofte en vigtig mulighed.
Ud over Medicin: Andre Vigtige Behandlingsaspekter
Medicinsk behandling er kun én del af en omfattende behandlingsplan. Andre elementer er lige så afgørende:
- Lungefysioterapi: Daglige øvelser og teknikker til at løsne og fjerne slim fra lungerne. Dette kan omfatte klapning, vibrationer (manuelt eller med en vest) og specifikke hoste- og vejrtrækningsteknikker.
- Ernæring: Patienter med CF har ofte brug for en diæt med højt kalorie- og fedtindhold for at kompensere for dårlig optagelse af næringsstoffer. De fleste skal tage pankreasenzymer i kapselform til hvert måltid for at hjælpe med fordøjelsen. Vitamintilskud (A, D, E og K) er også nødvendige.
- Motion: Regelmæssig fysisk aktivitet er utrolig vigtigt for at styrke vejrtrækningsmusklerne, forbedre lungefunktionen og den generelle sundhed.
Ofte Stillede Spørgsmål (FAQ) om Cystisk Fibrose
Hvad betyder en positiv nyfødtscreening for CF?
En positiv screening betyder ikke automatisk, at dit barn har cystisk fibrose. Screeningen måler et stof i blodet, der kan være forhøjet af flere årsager. En endelig diagnose kræver en svedtest, som måler saltindholdet i sveden. De fleste babyer med en positiv screening viser sig heldigvis ikke at have CF.
Hvordan arves cystisk fibrose?
Sygdommen arves autosomalt recessivt. Det betyder, at et barn skal arve et defekt CFTR-gen fra begge forældre for at udvikle sygdommen. Hvis man kun arver ét defekt gen, er man en rask bærer af sygdommen. Hvis begge forældre er bærere, er der 25% chance for, at hvert barn vil blive født med cystisk fibrose.
Kan man blive diagnosticeret med CF som voksen?
Ja, næsten 10% af alle tilfælde diagnosticeres i voksenalderen. Dette kan skyldes, at nogle genmutationer forårsager en mildere form af sygdommen, hvor symptomerne først bliver tydelige senere i livet. Desuden blev nyfødtscreening for CF først standard i mange lande efter 2010.
Påvirker CF fertiliteten?
Ja, fertiliteten er påvirket hos både mænd og kvinder. Kvinder kan have tykkere slim i livmoderhalsen, hvilket kan gøre det sværere at blive gravid, men de fleste kan få børn. Næsten alle mænd med CF er infertile, fordi sædlederen mangler. De producerer dog normal sæd, og kan derfor få biologiske børn ved hjælp af assisteret reproduktionsteknologi.
Er cystisk fibrose den samme for alle?
Absolut ikke. Sygdommen varierer enormt fra person til person. Nogle har milde symptomer, der primært påvirker ét organ, mens andre har en alvorlig sygdom, der påvirker flere organsystemer. Den specifikke genmutation spiller en stor rolle, men et specialiseret CF-behandlingsteam vil altid udarbejde en personlig behandlingsplan for at imødekomme den enkeltes unikke behov.
Hvis du vil læse andre artikler, der ligner Behandling af Cystisk Fibrose: En Komplet Guide, kan du besøge kategorien Sundhed.