06/01/2001
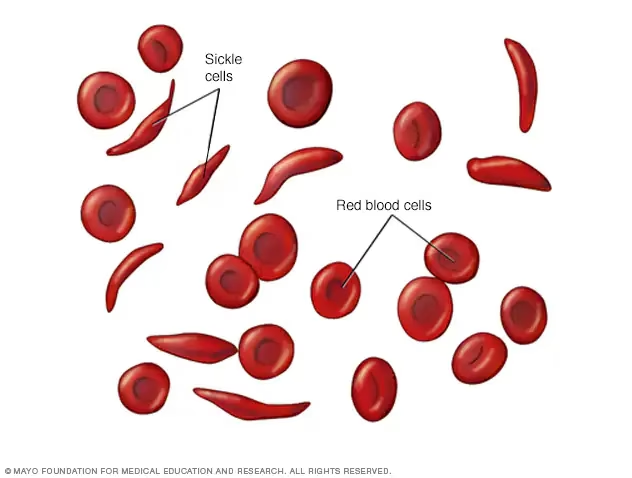
Seglcelleanæmi er en arvelig blodsygdom, der påvirker de røde blodlegemer, som er ansvarlige for at transportere ilt rundt i kroppen. Hos raske mennesker er de røde blodlegemer runde og fleksible, hvilket gør det let for dem at bevæge sig gennem selv de mindste blodkar. Men for en person med seglcelleanæmi er historien en helt anden. Sygdommen tvinger de røde blodlegemer til at antage en unormal, stiv segl- eller C-form, hvilket kan føre til en række alvorlige helbredsproblemer. Denne artikel vil udforske alle aspekter af seglcelleanæmi, fra dens genetiske oprindelse til de nyeste behandlingsstrategier.
Hvad er Seglcelleanæmi?
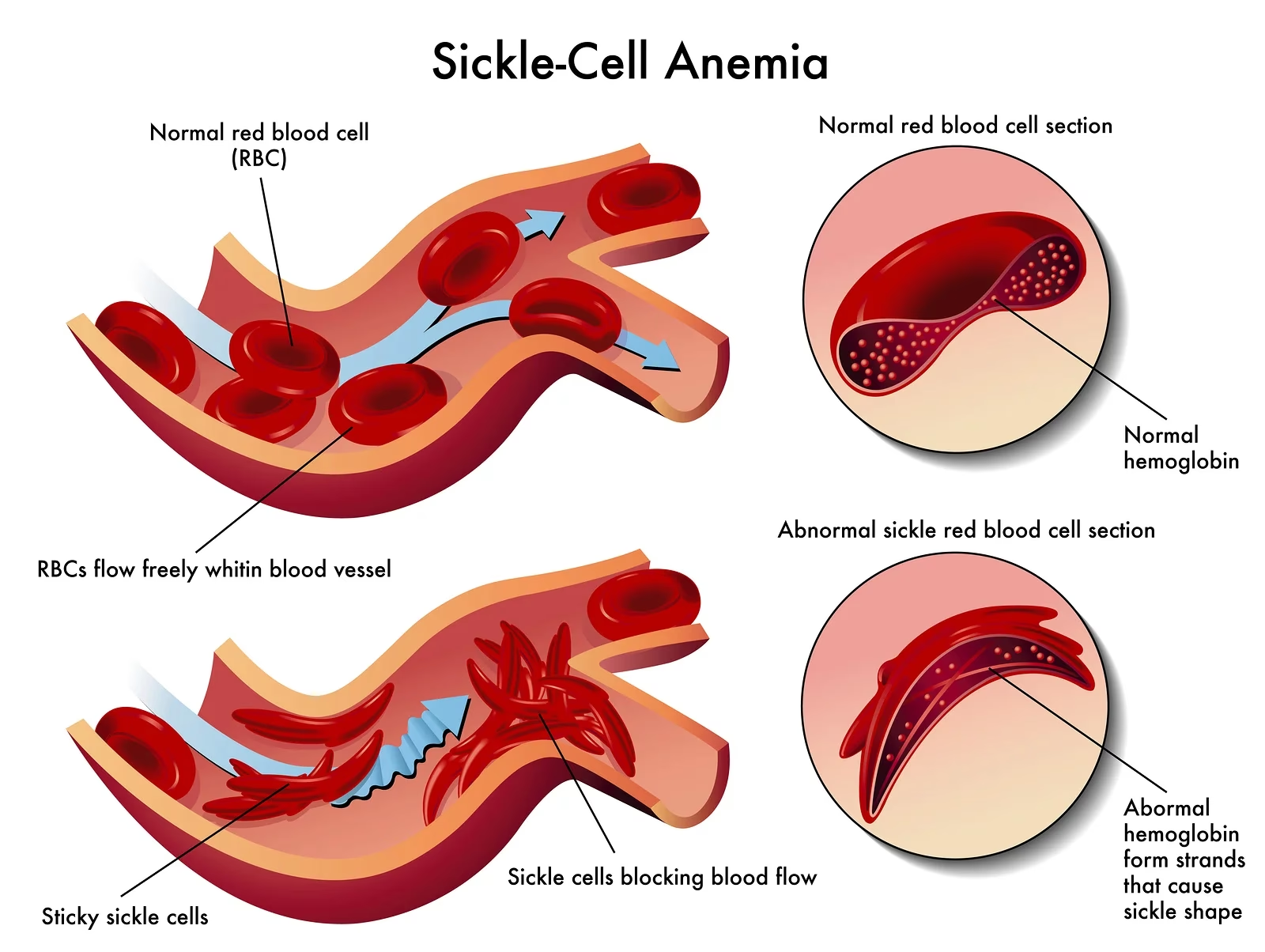
For at forstå seglcelleanæmi, må vi først se på de røde blodlegemers funktion. Disse celler indeholder et protein kaldet hæmoglobin, som binder ilt i lungerne og frigiver det i resten af kroppens væv. Ved seglcelleanæmi er der en defekt i det gen, der koder for hæmoglobin. Dette resulterer i produktionen af en unormal type hæmoglobin, kendt som hæmoglobin S (HbS).
Når hæmoglobin S frigiver ilt, får det de røde blodlegemer til at kollapse og blive hårde, klæbrige og formet som en segl. Disse seglceller har to store problemer:
- De dør for tidligt: Mens normale røde blodlegemer lever i omkring 120 dage, overlever seglceller typisk kun 10 til 20 dage. Kroppen kan ikke producere nye røde blodlegemer hurtigt nok til at erstatte dem, der dør, hvilket fører til en konstant mangel – en tilstand kendt som anæmi. Anæmi kan forårsage træthed, svimmelhed og åndenød.
- De blokerer blodgennemstrømningen: På grund af deres stive form og klæbrige overflade kan seglceller let sætte sig fast i små blodkar og blokere blodgennemstrømningen. Dette forhindrer ilt i at nå forskellige dele af kroppen, hvilket kan forårsage pludselige, intense smerter og over tid føre til alvorlig organskade.
Årsager: Genetikken bag Sygdommen
Seglcelleanæmi er en genetisk lidelse, man fødes med. Det er ikke smitsomt. Sygdommen nedarves på en autosomal recessiv måde, hvilket betyder, at et barn skal arve et defekt hæmoglobin-gen fra begge forældre for at udvikle sygdommen.
Hvis en person kun arver ét seglcelle-gen fra den ene forælder og et normalt hæmoglobin-gen (hæmoglobin A) fra den anden, har vedkommende det, der kaldes seglcelletræk (SCT eller HbAS). Personer med seglcelletræk er bærere af genet, men har typisk ingen symptomer på sygdommen. De kan dog give det defekte gen videre til deres børn. I sjældne tilfælde, under ekstreme forhold som dehydrering, høje højder eller intens fysisk anstrengelse, kan personer med seglcelletræk opleve komplikationer.
Forskellige Typer af Seglcelleanæmi
Der findes flere forskellige typer af seglcelleanæmi, afhængigt af hvilke hæmoglobin-gener en person arver. Sværhedsgraden af sygdommen varierer meget mellem de forskellige typer.
Sammenligning af de mest almindelige typer
| Type | Genetisk arvegang | Typisk sværhedsgrad |
|---|---|---|
| HbSS (Seglcelleanæmi) | Arver to hæmoglobin 'S' gener. | Dette er den mest almindelige og typisk den mest alvorlige form af sygdommen. |
| HbSC | Arver et 'S' gen og et 'C' gen (en anden type unormalt hæmoglobin). | Normalt en mildere form for seglcelleanæmi. |
| HbS beta-thalassæmi | Arver et 'S' gen og et gen for beta-thalassæmi. | Sværhedsgraden varierer. HbS beta0-thalassæmi er ofte lige så alvorlig som HbSS, mens HbS beta+-thalassæmi er mildere. |
| Seglcelletræk (HbAS) | Arver et 'S' gen og et normalt 'A' gen. | Ikke en sygdom, men en bærertilstand. Normalt ingen symptomer. |
Der findes også sjældnere typer som HbSD, HbSE og HbSO, hvor sværhedsgraden kan variere betydeligt.
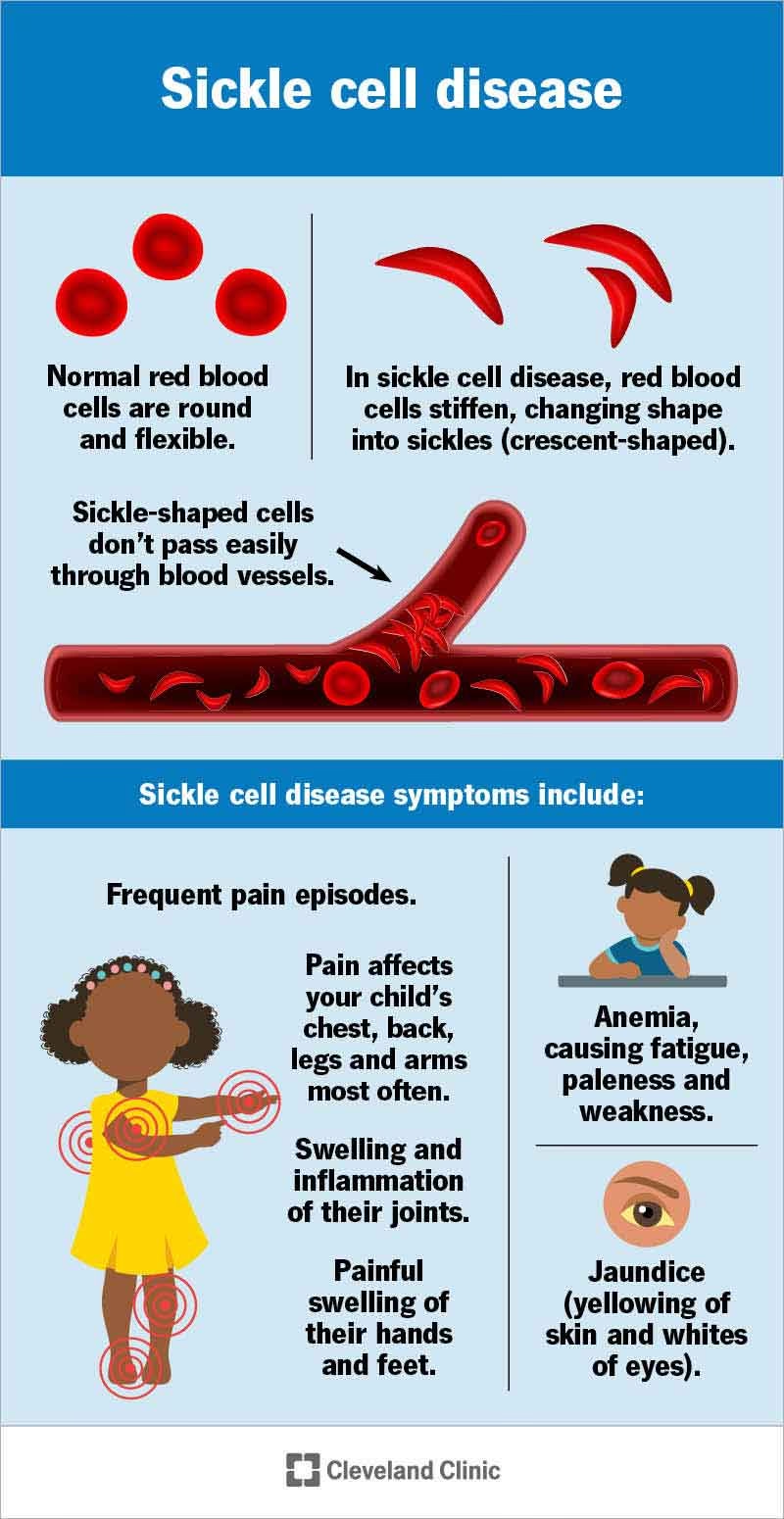
Symptomer og Komplikationer
Symptomer på seglcelleanæmi viser sig typisk i løbet af det første leveår, ofte omkring 5-månedersalderen. Symptomerne og komplikationerne kan variere fra milde til livstruende og er forskellige fra person til person.
- Smertefulde kriser: En af de mest almindelige og belastende komplikationer er episoder med intense smerter, kendt som smertefulde kriser eller vaso-okklusive kriser. De opstår, når seglceller blokerer blodgennemstrømningen til et bestemt område af kroppen, f.eks. brystet, maven, led eller knogler.
- Kronisk anæmi: Den konstante mangel på røde blodlegemer forårsager vedvarende træthed, svaghed og bleghed.
- Hævelse af hænder og fødder: Dette er ofte et af de første tegn på seglcelleanæmi hos spædbørn og skyldes, at seglceller blokerer blodkarrene i hænder og fødder.
- Hyppige infektioner: Seglcelleanæmi kan beskadige milten, et organ der er vigtigt for at bekæmpe infektioner. Dette gør især små børn sårbare over for alvorlige bakterielle infektioner.
- Akut brystsyndrom: En livstruende komplikation, der ligner lungebetændelse. Den skyldes, at seglceller blokerer blodkarrene i lungerne, og symptomerne omfatter brystsmerter, feber og åndedrætsbesvær.
- Slagtilfælde: Børn og voksne med seglcelleanæmi har en øget risiko for slagtilfælde, hvis seglceller blokerer blodkar i hjernen.
- Organskader: Over tid kan den nedsatte blod- og iltforsyning forårsage skader på organer som nyrer, lever og hjerte.
Diagnose og Screening
Seglcelleanæmi diagnosticeres ved hjælp af en simpel blodprøve. I mange lande, herunder USA og dele af Europa, er screening for seglcelleanæmi en standard del af de screeninger, nyfødte gennemgår på hospitalet. Dette er afgørende, da tidlig diagnose og behandling kan forhindre alvorlige komplikationer.
De specifikke laboratorietests, der bruges til at identificere unormalt hæmoglobin, omfatter:
- Højtydende væskekromatografi (HPLC)
- Hæmoglobin isoelektrisk fokusering (Hb IEF)
Diagnosen kan også stilles før fødslen ved hjælp af prænatale tests som moderkagebiopsi (chorionic villus sampling) eller fostervandsprøve (amniocentesis), hvor man analyserer fosterets genetiske materiale.
Behandlingsmuligheder og Håndtering
Selvom seglcelleanæmi er en livslang sygdom, har behandlingsmulighederne forbedret sig markant, hvilket har øget både livskvalitet og forventet levetid for patienter. Behandlingen fokuserer på at kontrollere symptomer og forebygge komplikationer.
Behandlingsstrategier:
- Smertehåndtering: Behandling af smertefulde kriser er en hjørnesten i behandlingen. Dette kan omfatte håndkøbsmedicin, receptpligtig smertestillende medicin og i alvorlige tilfælde hospitalsindlæggelse.
- Forebyggelse af infektioner: Børn med seglcelleanæmi får ofte dagligt penicillin indtil 5-årsalderen for at forebygge livstruende infektioner. Det er også afgørende, at de modtager alle anbefalede vaccinationer.
- Medicin: Lægemidler som hydroxyurea kan reducere hyppigheden af smertefulde kriser og akut brystsyndrom ved at øge produktionen af føtalt hæmoglobin (HbF), en type hæmoglobin, der forhindrer segldannelse.
- Blodtransfusioner: Regelmæssige blodtransfusioner kan behandle alvorlig anæmi og hjælpe med at forebygge slagtilfælde hos børn med høj risiko.
- Stamcelletransplantation: Også kendt som knoglemarvstransplantation, er dette den eneste potentielle kur mod seglcelleanæmi. Proceduren indebærer at erstatte patientens knoglemarv med sund knoglemarv fra en matchende donor (ofte en søskende). Det er dog en risikabel procedure og er ikke en mulighed for alle.
Ofte Stillede Spørgsmål (FAQ)
Er seglcelleanæmi smitsomt?
Nej, absolut ikke. Seglcelleanæmi er en arvelig, genetisk sygdom, som man fødes med. Man kan ikke "få" den fra en anden person, som man kan med en forkølelse eller influenza.
Kan man blive helbredt for seglcelleanæmi?
For de fleste er seglcelleanæmi en livslang tilstand, der kræver løbende behandling og håndtering. Den eneste nuværende potentielle kur er en stamcelle- eller knoglemarvstransplantation, men denne procedure er forbundet med betydelige risici og kræver en tæt matchende donor.
Hvad er forskellen på seglcelleanæmi og seglcelletræk?
Seglcelleanæmi (f.eks. type HbSS) er selve sygdommen, hvor en person har arvet to seglcelle-gener og oplever symptomer. Seglcelletræk (HbAS) er en bærertilstand, hvor en person har arvet ét seglcelle-gen og ét normalt gen. Personer med seglcelletræk er generelt raske, men kan give genet videre til deres børn.
Hvorfor kaldes det "seglcelle"?
Navnet kommer fra cellernes form. Under visse forhold ændrer de røde blodlegemer form fra en rund disk til en halvmåne- eller C-form, der ligner en landbrugsredskab kaldet en segl.
Hvis du vil læse andre artikler, der ligner Seglcelleanæmi: Alt du behøver at vide, kan du besøge kategorien Sundhed.