18/10/2004
Med indførelsen af EU's forordning om medicinsk udstyr (MDR 2017/745) er der kommet et markant øget fokus på sikkerhed, kvalitet og sporbarhed for medicinsk udstyr på det europæiske marked. En central del af denne forordning er den formelle definition og tildeling af ansvar til de såkaldte økonomiske operatører. Disse aktører udgør rygraden i forsyningskæden, og forordningen sikrer, at hver enkelt part bærer et juridisk ansvar for, at udstyret lever op til de strenge krav. Dette skift betyder, at ansvaret ikke længere udelukkende hviler på producenten, men deles på tværs af hele kæden for at skabe et mere robust sikkerhedsnet for patienter og brugere.

Formålet er at implementere kontrolmekanismer i hvert led af processen, fra design og fremstilling til distribution og service. Hver økonomisk operatør fungerer som en kontrolpost for de andre, hvilket skaber en kæde af gensidig ansvarlighed. Denne artikel dykker ned i, hvem disse økonomiske operatører er, og hvilke specifikke ansvarsområder de har under MDR.
Hvem er de Økonomiske Operatører ifølge MDR?
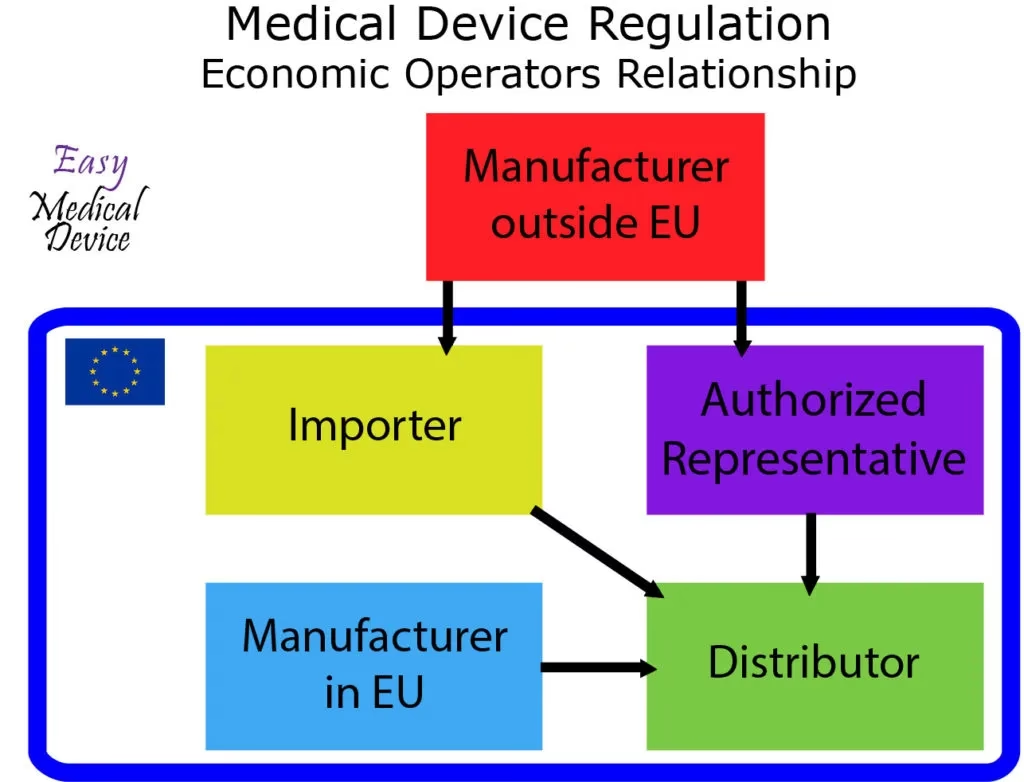
Artikel 2 i EU's MDR og IVDR (Forordning om medicinsk udstyr til in vitro-diagnostik) definerer fire forskellige roller, der formelt klassificeres som 'økonomiske operatører'. Disse er:
- Producent: En fysisk eller juridisk person, der fremstiller eller fuldstændigt renoverer et stykke udstyr, eller får et stykke udstyr designet, fremstillet eller fuldstændigt renoveret, og markedsfører dette udstyr under eget navn eller varemærke.
- Autoriseret Repræsentant: En fysisk eller juridisk person, der er etableret i Unionen, og som skriftligt har modtaget og accepteret et mandat fra en producent, der er beliggende uden for Unionen, til at handle på producentens vegne i forbindelse med specifikke opgaver i henhold til denne forordnings forpligtelser.
- Importør: En fysisk eller juridisk person, der er etableret i Unionen, og som bringer et stykke udstyr fra et tredjeland i omsætning på EU-markedet.
- Distributør: En fysisk eller juridisk person i forsyningskæden, bortset fra producenten eller importøren, der gør et stykke udstyr tilgængeligt på markedet, indtil det tages i brug.
Under MDR og IVDR deler disse fire operatører ansvaret for at sikre overholdelse af reglerne. Dette princip om 'solidarisk ansvar' er en af de mest betydningsfulde ændringer i den nye lovgivning og har til formål at styrke markedsovervågningen og patientsikkerheden.
Producentens Omfattende Ansvar
Producenten er kernen i det regulatoriske system og bærer det primære ansvar for sit medicinske udstyrs overensstemmelse med lovgivningen. Deres forpligtelser er de mest omfattende og inkluderer:
- CE-mærkning: Sikre, at udstyret opfylder de væsentlige sikkerheds- og ydeevnekrav i MDR's Bilag I og påfører CE-mærkning som bevis herpå.
- Teknisk Dokumentation: Udarbejde og vedligeholde en omfattende teknisk dokumentation i overensstemmelse med Bilag II og III, som demonstrerer udstyrets overensstemmelse. Dette omfatter risikoanalyse, klinisk evaluering og testdata.
- Kvalitetsstyringssystem (QMS): Implementere og vedligeholde et effektivt QMS, der som minimum lever op til kravene i ISO 13485 og MDR's Bilag IX.
- Overvågning efter Markedsføring (PMS): Etablere en proaktiv PMS-plan for løbende at overvåge udstyrets ydeevne og sikkerhed, når det er på markedet.
- Hændelsesrapportering: Rapportere alvorlige hændelser og korrigerende sikkerhedsforanstaltninger på markedet (FSCA) til de relevante kompetente myndigheder.
- Unik Produktidentifikation (UDI): Tildele og vedligeholde UDI'er for alt udstyr for at sikre fuld sporbarhed gennem hele forsyningskæden.
- Dokumentopbevaring: Opbevare den tekniske dokumentation, EU-overensstemmelseserklæringen og relevante certifikater i mindst 10 år (15 år for implantater) efter, at det sidste produkt er bragt i omsætning.
- Økonomisk Dækning: Sikre tilstrækkelig økonomisk dækning til at dække potentielt ansvar i henhold til direktivet om produktansvar (85/374/EØF), proportionalt med risikoklassen og virksomhedens størrelse.
Den Autoriserede Repræsentants Vigtige Rolle
For producenter uden for EU er den autoriserede repræsentant (AR) en uundværlig partner og det primære kontaktpunkt til de europæiske myndigheder. AR'en er ikke blot en postadresse, men en juridisk ansvarlig enhed.
Nøgleansvarsområder for AR:
- Verifikation af Overholdelse: AR skal verificere, at producenten har udarbejdet den nødvendige tekniske dokumentation og EU-overensstemmelseserklæring, samt har gennemført de korrekte overensstemmelsesvurderingsprocedurer.
- Dokumentadgang: AR skal opbevare en kopi af den tekniske dokumentation, erklæringer og certifikater og gøre dem tilgængelige for kompetente myndigheder på anmodning.
- Kommunikation: Fungerer som den primære kommunikationskanal mellem producenten og de nationale myndigheder samt bemyndigede organer.
- Vigilans og Hændelsesrapportering: AR skal straks informere producenten om klager og rapporter om hændelser modtaget fra sundhedspersonale, patienter eller brugere. De skal også samarbejde om implementering af korrigerende handlinger.
- Registrering: Sikre at producenten og udstyret er korrekt registreret i den europæiske database for medicinsk udstyr, EUDAMED.
- Juridisk Ansvar: AR kan holdes solidarisk ansvarlig sammen med producenten for defekt udstyr, hvis producenten, der er baseret uden for EU, ikke opfylder sine forpligtelser.
Importørens Kontrolfunktion
Importører har fået en markant udvidet rolle under MDR. De er ikke længere blot logistiske mellemled, men en aktiv kontrolinstans, der skal sikre, at udstyr fra tredjelande er i fuld overensstemmelse med EU-lovgivningen, før det kommer på markedet.
Importørens Ansvarsliste:
- Verifikation før Import: Før et produkt importeres, skal importøren verificere, at det er CE-mærket, at der er tildelt en UDI, at der er en EU-overensstemmelseserklæring, og at producenten har udpeget en autoriseret repræsentant.
- Identifikation: Importøren skal anføre sit eget navn, registrerede firmanavn eller varemærke samt adresse på udstyret, emballagen eller i et medfølgende dokument. Dette må ikke skjule producentens oplysninger.
- Lager- og Transportbetingelser: Sikre, at opbevarings- og transportforholdene, mens udstyret er under deres ansvar, ikke kompromitterer dets sikkerhed og ydeevne og er i overensstemmelse med producentens anvisninger.
- Klagesystem: Føre et register over klager, ikke-overensstemmende udstyr, tilbagekaldelser og tilbagetrækninger og dele relevant information med producenten, AR og distributører.
- Myndighedssamarbejde: Samarbejde med kompetente myndigheder og på anmodning fremlægge dokumentation eller gratis vareprøver.
Distributørens Ansvar i Sidste Led
Distributører er ofte det sidste led i kæden, inden udstyret når hospitaler, klinikker eller patienter. MDR pålægger dem en pligt til at handle med 'rettidig omhu' for at sikre, at kun sikkert og overensstemmende udstyr gøres tilgængeligt.
Distributørens Forpligtelser:
- Verifikation før Distribution: Ligesom importøren skal distributøren verificere, at udstyret er CE-mærket, at det ledsages af den nødvendige information (f.eks. brugsanvisning på det korrekte sprog), og at importøren (hvis relevant) har opfyldt sine mærkningskrav.
- Sporbarhed: Skal kunne identificere enhver økonomisk operatør, som de har fået leveret udstyr fra, og som de har leveret udstyr til.
- Håndtering af Udstyr: Sikre, at lager- og transportbetingelser overholder producentens anvisninger for at undgå beskadigelse eller forringelse af udstyret.
- Videresendelse af Information: Skal øjeblikkeligt videresende klager eller rapporter om formodede hændelser til producenten (og eventuelt AR og importør).
- Korrektive Handlinger: Samarbejde med producenten og myndighederne om at trække udstyr tilbage eller tilbagekalde det, hvis det viser sig at være ikke-overensstemmende.
Sammenligning af Importør og Distributør
Selvom deres roller kan virke ens, er der vigtige forskelle i deres ansvarsområder, især hvad angår det første skridt for at bringe et produkt ind på EU-markedet.
| Opgave | Importør | Distributør |
|---|---|---|
| Primær Funktion | Bringer udstyr fra et land uden for EU ind på EU-markedet. | Gør udstyr, der allerede er på EU-markedet, tilgængeligt i forsyningskæden. |
| Identifikation på Produkt | Skal tilføje egne kontaktoplysninger på produkt/emballage. | Skal ikke tilføje egne oplysninger, men skal verificere importørens oplysninger. |
| Registrering i EUDAMED | Skal registrere sig selv i EUDAMED. | Skal ikke registrere sig i EUDAMED. |
| Verifikation af AR | Skal verificere, at en autoriseret repræsentant er udpeget. | Indirekte verifikation ved at tjekke importørens overholdelse. |
Ofte Stillede Spørgsmål
Hvem er de fire økonomiske operatører under MDR?
De fire økonomiske operatører er producenten, den autoriserede repræsentant, importøren og distributøren. Hver har specifikke juridiske forpligtelser under forordningen.
Hvad er den største ændring for importører og distributører?
Den største ændring er skiftet fra at være passive led i forsyningskæden til at have et aktivt og juridisk ansvar for at verificere udstyrets overensstemmelse. De kan holdes ansvarlige for at markedsføre udstyr, der ikke lever op til kravene, og skal derfor have systemer på plads til kontrol og dokumentation.
Hvad betyder 'solidarisk ansvar' i praksis?
Solidarisk ansvar (joint and several liability) betyder, at flere parter kan holdes ansvarlige for den samme skade. For eksempel kan en patient, der lider skade på grund af et defekt produkt, rette sit erstatningskrav mod både producenten og den autoriserede repræsentant eller importøren. Dette øger incitamentet for alle parter til at sikre, at reglerne overholdes.
Hvorfor skal en autoriseret repræsentant udpeges?
En autoriseret repræsentant er påkrævet for alle producenter, der er baseret uden for EU, men som ønsker at sælge deres medicinske udstyr på EU-markedet. Repræsentanten fungerer som en juridisk enhed inden for EU, som myndighederne kan henvende sig til, og som deler ansvaret for produktets sikkerhed.
Konklusion: Et Fælles Løft for Patientsikkerheden
EU's forordning om medicinsk udstyr har fundamentalt ændret landskabet for alle, der er involveret i forsyningskæden for medicinsk udstyr. Ved klart at definere rollerne og ansvarsområderne for de økonomiske operatører – producent, autoriseret repræsentant, importør og distributør – har MDR skabt et system, hvor ansvaret deles, og hvor hver enkelt aktør spiller en afgørende rolle i at sikre, at kun sikkert og effektivt udstyr når frem til patienterne. Denne model med delt ansvar er designet til at styrke overvågningen, øge gennemsigtigheden og i sidste ende forbedre folkesundheden i hele Europa.
Hvis du vil læse andre artikler, der ligner MDR: De Økonomiske Operatørers Ansvarsroller, kan du besøge kategorien Sundhed.